KEYWORDS
Dapsone, methaemoglobinaemia, methylene blue, rebound
CASE DESCRIPTION
A 28-year-old woman visited the Emergency Department of our hospital with acute hypoxic respiratory distress. The patient was experiencing progressive dyspnoea for a couple of days and had experienced multiple fainting spells. On examination, she had an oxygen saturation measured with pulse oximeter of 80-85% (SpO2), and central and peripheral cyanosis, which did not improve with supplementation of 15 litres of oxygen/minute using a non-rebreather mask. Because of her condition, the patient was immediately intubated and ventilated with pressure support. Unfortunately, SpO2 also did not improve despite maximum fraction of inspired oxygen (FiO2) of 100%. A CT scan ruled out pulmonary embolism and intraparenchymatous abnormalities explaining the low saturation. Transthoracic ultrasound showed normal ventricular function and no signs of cardiac shunting. Arterial blood samples were drawn, which had a striking chocolate brown colour, and blood gas analysis showed a normal oxygen saturation of 98%, in comparison with the measured 85% with pulse-oximetry. Surprisingly, blood gas analysis showed that the methaemoglobin (MetHb) level was 21.6% (0.216 mol/mol), almost 15 times higher than the cut-off normal value of 1.5% (0.015 mol/mol).
After initial stabilisation and treatment, additional information about her medical history became clear. She was diagnosed with coeliac disease and was also suspected of dermatitis herpetiformis, for which the antibiotic dapsone was prescribed and dosage was increased from 50 to 100 mg, once daily, four days prior to her visit to the Emergency Department. Based on these results, the most likely diagnosis was thought to be symptomatic methaemoglobinaemia caused by dapsone. Methylene blue was given intravenously, after which her MetHb level quickly declined (figure 1) and shortly after infusion, she was extubated because of substantial clinical improvement, in particular, the normalisation of her blood oxygenation. Dapsone was discontinued. 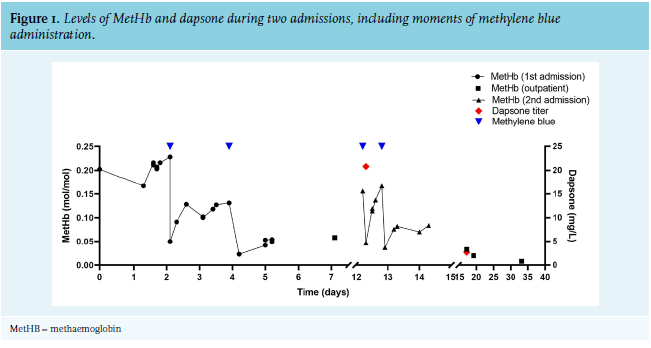
 Surprisingly, seven days after discharge and 12 days after discontinuing dapsone, the patient presented again with symptoms of progressive dyspnoea, which appeared to be based on a recurrent symptomatic methaemoglobinaemia. MetHb level was 15.6% (0.156 mol/mol), a 3-fold increase compared to the levels at time of discharge. The patient was readmitted and again treated with methylene blue, and after two doses, her MetHb level stayed within normal ranges (figure 1). Toxicological analysis of a blood sample taken 12 days after discontinuation of dapsone showed a dapsone concentration of 20.8 mg/l, which is within the toxic range since the reference therapeutic concentrations are between 0.5-2.0 mg/l. Unfortunately, no toxicological analysis was performed on blood samples taken during the first episode. The patient assured us she had not taken any dapsone, or any other medication or drugs of abuse such as ‘poppers’, which was confirmed by toxicological screening. Her pharmacy confirmed that she had turned in the remaining tablets after discharge from the previous admission; furthermore, she used no medication influencing pharmacokinetics of dapsone. The toxicological analysis supports our theory that dapsone is the most likely agent for the relapse of the methaemoglobinaemia with a score of 7, as assessed by the Naranjo algorithm for assessment of adverse drug reactions.1,2 Due to the toxic concentration, we hypothesised that the elimination of dapsone was delayed or that there was a rebound.
Surprisingly, seven days after discharge and 12 days after discontinuing dapsone, the patient presented again with symptoms of progressive dyspnoea, which appeared to be based on a recurrent symptomatic methaemoglobinaemia. MetHb level was 15.6% (0.156 mol/mol), a 3-fold increase compared to the levels at time of discharge. The patient was readmitted and again treated with methylene blue, and after two doses, her MetHb level stayed within normal ranges (figure 1). Toxicological analysis of a blood sample taken 12 days after discontinuation of dapsone showed a dapsone concentration of 20.8 mg/l, which is within the toxic range since the reference therapeutic concentrations are between 0.5-2.0 mg/l. Unfortunately, no toxicological analysis was performed on blood samples taken during the first episode. The patient assured us she had not taken any dapsone, or any other medication or drugs of abuse such as ‘poppers’, which was confirmed by toxicological screening. Her pharmacy confirmed that she had turned in the remaining tablets after discharge from the previous admission; furthermore, she used no medication influencing pharmacokinetics of dapsone. The toxicological analysis supports our theory that dapsone is the most likely agent for the relapse of the methaemoglobinaemia with a score of 7, as assessed by the Naranjo algorithm for assessment of adverse drug reactions.1,2 Due to the toxic concentration, we hypothesised that the elimination of dapsone was delayed or that there was a rebound.
Based on literature which will be reviewed later, cimetidine and ascorbic acid were added to the treatment to prevent a new relapse and to further diminish MetHb levels. Two days after readmission, the patient could be discharged from the hospital free of symptoms. Frequent outpatient clinic follow-up showed no increase of MetHb levels thereafter, and the concentration of dapsone decreased to 2.7 mg/l in five days. Since then, there has been no more relapse of symptomatic methaemoglobinaemia (figure 1).
Methaemoglobinaemia by dapsone
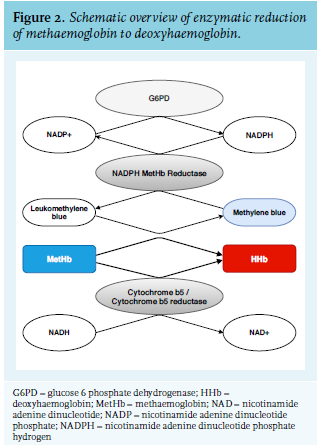
Dapsone is an antimicrobial agent registered for the treatment of leprosy and dermatitis herpetiformis. It can sometimes be used for other inflammatory conditions or as prophylaxis for pneumocystis jiroveci pneumonitis on an off-label basis. Serious side effects are rare but include agranulocytosis, methaemoglobinaemia, and haemolysis, especially in patients with G6PD-deficiency.3 Methaemoglobinaemia is a congenital or acquired functional anaemia in which the ferrous iron (iron(II) or Fe2+) of haemoglobin is oxidised to ferric iron (iron(III) or Fe3+). Under normal conditions, iron is reduced to Fe2+ to maintain a MetHb level of < 1%. This reduction takes place by two pathways involving nonenzymatic antioxidants: the cytochrome b5 and NADPH reductase pathways (figure 2). Medication and toxins are the major cause of acquired methaemoglobinaemia. Dapsone, chloroquine, primaquine, and local anaesthetics such as benzocaine and lidocaine are most common; the latter, even if administered topically, can cause significant methaemoglobinaemia. Chemicals known to cause methemoglobinemia are anilines, hydrogen peroxide, chlorates, and nitrates, ranging from polluted water and high-nitrate food to party drugs and medicinal nitrates.4 In dapsone, the toxic metabolite hydroxylamine is responsible for oxidising haemoglobin and thus the formation of MetHb.5
Oxidised iron in haem is not capable of dissociating oxygen because it changes the conformation of the remaining haemoglobin, resulting in a higher oxygen (O2) affinity. The oxygen dissociation curve shows a left shift and O2 delivery to the tissues is decreased, leading to cellular hypoxia and cyanosis. Symptoms vary based on MetHb concentration, ranging from asymptomatic cyanosis and headache to coma and severe hypoxic symptoms, as summarised in table 1.6-9
Clinical clues of methaemoglobinaemia are chocolate brown blood, no improvement of blood oxygen saturation measured by pulse-oximetry despite supplementation of oxygen with 100% FiO2 and a so called ‘saturation gap’: a difference between saturation measured with pulse-oximetry and blood gas analyser.
A simple bedside test can distinguish deoxyhaemoglobin from MetHb: Blood containing concentrations of MetHb of 20% and larger can appear chocolate brown; the colour gets darker with an increase in MetHb concentrations. When blood is exposed to atmospheric oxygen, for example, in cases of MetHb, its appearance does not change in contrast to deoxyhaemoglobin, in which its colour changes to bright red. This test is used in low resource settings to establish methaemoglobinaemia; it is also used to estimate the percentage of MetHb.10
Pulse oximeters measure the ratio of absorbance from pulsatile light transmission through vascular beds at two wavelengths, typically 660 and 940 nm, based upon the spectral properties of oxyhaemoglobin and deoxyhaemoglobin. The spectral properties of MetHb cause an underestimation of O2 saturation by pulse oximetry in increasing MetHb levels and eventually a stable plateau of approximately 85% saturation.9,11 Additionally, after administration of methylene blue, it becomes impossible to measure O2 saturation by pulse oximetry since methylene blue also absorbs light at the same wavelengths, and is not confined to the vascular system as it is also present in other tissues. However, some pulse oximeters use multiple wavelengths and can discriminate methaemoglobin, such as the Rad-57 Pulse CO-Oximeter (Masimo, Switzerland).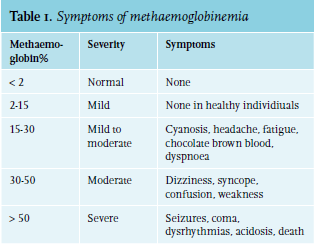
Blood gas analysis
While pulse oximetry suffers from interference by MetHb and methylene blue, the spectrophotometry-based blood gas analysis by modern analysers is accurate. Unfortunately, there still are misunderstandings about the interference of methylene blue in the measurement of the MetHb concentration.9,12
Previously, MetHb was determined from the absorption measured at two wavelengths, analogously to modern pulse oximetry, while cyanide was used to distinguish MetHb from other haemoglobin (Hb) derivatives (Evelyn-Malloy method).13 Later, carbon monoxide (CO)-oximetry methods were developed using four or more wavelengths, theoretically enabling the simultaneous determination of oxygenated Hb (O2Hb), deoxygenated Hb (HHb), carboxyhaemoglobin (COHb), and MetHb. Unfortunately, various CO-oximetry methods using 4 or up to 17 wavelengths are reported to yield inaccurate MetHb measurements in the presence of methylene blue.12,14 Today however, using modern analysers, the interference of methylene blue on MetHb measurement is negligible. For example, the ABL90 Flex Plus (Radiometer, Denmark), utilises CO oximetry with a 256-wavelength spectrophotometer in the range of 467-672 nm. The theoretical spectra of O2Hb, HHb, COHb, MetHb, fetal Hb (HbF), and bilirubin are fitted to the measured spectrum from the patient’s sample, enabling the separate determination of the concentration of these six parameters. Due to the large number of wavelengths used to determine the concentrations, the influence of interference is
minimised.15 Therefore, MetHb results obtained using this blood gas analyser are correct and may be used safely in clinical practice.
TREATMENT
Treatment is indicated in cases where MetHb concentrations are > 20% or in cases with symptoms of impaired tissue oxygenation. The first reference to methylene blue as a treatment of methaemoglobinaemia is from the early 1930s,16 but no clinical trials were published. Methylene blue is a cofactor of the nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) reductase pathway and forms leucomethylene blue, which acts as an electron donor and reduces MetHb to Hb (figure 2). The usual dose is 1-2 mg/kg in a 1% solution, intravenously administered over 5 to 10 minutes. The effect is expected in minutes to one hour. Treatment can be repeated to a maximum of 4 mg/kg/day to prevent toxicity, mainly paradoxical (mild) haemolysis.17,18
Methylene blue is ineffective in patients with glucose6-phosphate dehydrogenase (G6PD) deficiency and can even cause severe hemolysis.19 G6PD is a rate-limiting enzyme of the pentose phosphate pathway, which reduces nicotinamide adenine dinucleotide phosphate (NADP) to NADPH; therefore in patients with G6PD deficiency, there are low levels of NADPH. Insufficient levels of NADPH fail to reduce methylene blue to its active metabolite leucomethylene blue and therefore cannot reduce MetHb to Hb. Methylene blue can induce G6PD-related haemolysis due to the formation of free radicals and direct oxidative stress, for which G6PD-deficient erythrocytes are more vulnerable. In cases of G6PD deficiency, ascorbic acid can be used at high doses up to four grams per day, which acts as a nonenzymatic MetHb-reducing agent. In addition, the hydrogen (H2) antagonist cimetidine can be added in a daily dose of 400 mg once. Cimetidine reduces the activity of several cytochrome P-450 enzymes, which are responsible for the formation of dapsone’s toxic metabolite hydroxylamine, and therefore lowers the formation of hydroxylamine. In extreme cases of methaemoglobinaemia, exchange transfusion must be considered, particularly in children.8,20
REASONS FOR REBOUND
To explain the relapse in our patient after the first discharge with normal MetHb levels, we performed a literature search and investigated several options. First, delayed renal clearance was excluded since her kidney function was normal (Chronic Kidney Disease-Epidemiology Collaboration (CKD-EPI) 107 ml/ min). Second, G6PD deficiency was unlikely due to the significant drop in MetHb concentration after methylene blue administration. In cases of G6PD deficiency, methylene blue would not have this effect because G6PD is essential for the production of NADPH, which is responsible for the reduction of Fe3+ to Fe2+. 19 The absence of G6PD deficiency was confirmed by determining the G6PD activity in our patient (172 U/mmol Hb; reference interval 82-130 U/mmol Hb). Third, to rule out delayed metabolism due to pharmacogenetic variants, the enzymatic pathways responsible for the metabolisation of dapsone were analysed through assessing genetic variants of the specific enzymes. Dapsone is acetylated by N-acetyltransferase 2 (NAT2) to the inactive metabolite monoacetyldapsone and will thereafter be deacetylated. Further metabolism is through CYP3A4 and CYP2C9. The toxic metabolite hydroxylamine, is obtained by CYP2C9 and CYP2C19. Pharmacogenetic analysis revealed that there were no deficiencies in these enzymes: the patient was an intermediate metaboliser for CYP2C9 (*1/*2) and NAT2 (*4/ *5), and a normal metaboliser for CYP3A4 (*1/*1). Therefore, we excluded delayed metabolism of dapsone as cause of recurring methaemoglobinaemia.
Furthermore, we found several theoretical explanations in earlier case reports also describing the recurrence of methaemoglobinaemia after the cessation of drug intake. First, dapsone has lipophilic characteristics (Vd = 1 l/kg and Log P= 0,97) and will therefore mostly be found in peripheral tissues.21 It has been hypothesised that dapsone concentrations in blood could be increased for a prolonged period as a result of the distribution of dapsone from the peripheral tissue to the blood.22 Additionally, dapsone undergoes enterohepatic circulation, which is responsible for high concentrations in the tissue even after three weeks of discontinuation; however, it is highly unlikely that this mechanism can be responsible for the toxic levels found in our case since most of the elimination of dapsone metabolites is in the urine.23 Moreover, once dapsone is converted into hydroxylamine and absorbed by erythrocytes, it can be recycled and converted back into dapsone. This recycling pathway may be responsible for the persistence of dapsone in the blood and thus the relapse of methaemoglobinaemia.24
A possibility that should always be considered is re-initiation of dapsone, possibly as part of Munchausen syndrome. However, the patient denied taking any additional dapsone after the first hospital admission and disposal of the remaining dosages was confirmed by her pharmacy.
CONCLUSION
We gave an overview of the pathophysiology; diagnostics, including potential interference of methylene blue with measurement of MetHb in modern blood gas analysers; and treatment of methaemoglobinaemia caused by dapsone. This case shows the importance of considering the possibility of a late rebound methaemoglobinaemia after discontinuation of dapsone and possible pathophysiological explanations for this phenomenon.
DISCLOSURES
All authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES