KEY WORDS
Diabetes insipidus, hypothalamic-pituitary axis, Langerhans cell histiocytosis
INTRODUCTION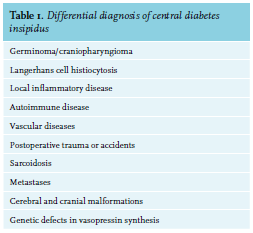
Diabetes insipidus (DI) is a disorder of water balance in the kidneys, characterized by a failure to concentrate urine, causing polyuria (i.e. > 2 l/m2 /24h) and subsequently polydipsia. DI is subdivided into central and nephrogenic DI. Central diabetes insipidus (CDI) arises from destruction or degeneration of magnocellular neurons in the paraventricular and supraoptic nuclei, which are responsible for producing vasopressin. If more than 80% of these cell bodies are damaged, polyuria occurs because of vasopressin deficiency. In contrast, nephrogenic DI is the result of an impaired kidney response to vasopressin. The broad differential diagnosis of CDI makes it challenging to determine its underlying aetiology (table 1).1 One cause of CDI is Langerhans cell histiocytosis (LCH), which can appear with variable presentation. We demonstrate this by describing three different patients with LCH.
SUBJECTS AND METHODS
Three cases of LCH with CDI were retrospectively analysed in the outpatient clinic of the University Hospital in Brussels from 1996 until 2017. Patient characteristics, clinical presentation, diagnosis and treatment were compared (table 2). 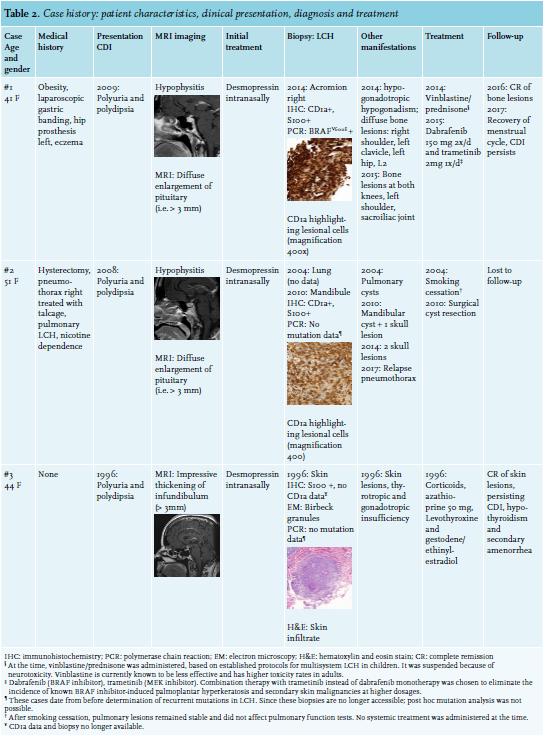
RESULTS
Case histories
Case 1: In 2009, a 41-year-old woman presented with polyuria and polydipsia. A water deprivation test confirmed the diagnosis of CDI, and magnetic resonance imaging (MRI) of the pituitary gland showed hypophysitis. Desmopressin treatment was started. In 2014, she developed skeletal pain and a fever. Positron emission tomography with 2-deoxy-2-fluorine-18-fluoro-D-glucose and computed tomography (18-FDG PET/CT) showed various bone lesions. Histological examination of lesional tissue at the right acromion showed positive CD1a and protein S100 staining; polymerase chain reaction (PCR) demonstrated a proto-oncogene BRAFV600E mutation matching the diagnosis of LCH. In the meantime, the patient also developed hypogonadotropic hypogonadism. Systemic treatment with vinblastine and prednisone was initiated, although suspended after six weeks because of toxicity. In 2015, new bone lesions were identified by 18-FDG PET/CT. Treatment with dabrafenib and trametinib was started until last follow-up, resulting in a complete remission of the skeletal lesions and recovery of the menstrual cycle. Desmopressin treatment has been continued because CDI persists.
Case 2: A 51-year-old woman with known pulmonary histiocytosis since 2004 developed polyuria and polydipsia in 2008. CDI was confirmed through a positive water deprivation test, and MRI of the pituitary gland demonstrated hypophysitis. Desmopressin treatment relieved her symptoms. A diagnostic technetium bone scintigraphy revealed lesions at the mandibula and skull. LCH was confirmed upon histological examination of the mandibular lesion expressing CD1a and protein S100 antigens. At the time, mutational analysis was not routinely performed to diagnose LCH. Therefore, we do not have any mutational data of the lesional tissue. The bone lesions were resected and although systemic treatment was repeatedly proposed because of multisystemic disease, the patient declined this.
Case 3: A 44-year-old woman presented in 1996 with polyuria and polydipsia. A water deprivation test confirmed the diagnosis of CDI, and MRI of the pituitary gland showed an impressive thickening of the infundibulum. Simultaneously, coexisting hypogonadotropic hypogonadism and central hypothyroidism were diagnosed. Desmopressin treatment and substitution therapy with levothyroxine and an oral contraceptive pill achieved good results. A few months later, she developed skin lesions. Underlying LCH was identified through electron microscopy and histological examination of a skin biopsy, showing typical Birbeck granules and protein S100 positivity, respectively. No mutational data are available for this case either. Corticosteroids and azathioprine treatment caused the lesions to disappear. Nevertheless, it was necessary to continue treatment for CDI and anterior pituitary function loss.
DISCUSSION
The above-described three patients demonstrate the highly variable presentation of LCH, in which the common denominator is the development of CDI.
LCH is a rare disease and can occur at any age, although it is more frequently seen in children with a male predominance.2 Its incidence is approximately five cases per million children and about one or two cases per million adults.3 General clinical presentation of LCH can be highly variable depending on which organ is infiltrated, and the disease spectrum ranges from isolated skin or skeletal lesions to multisystem involvement and sometimes life-threatening conditions such as acute lymphoblastic leukaemia.4 A four-part LCH classification has been established by the Histiocyte Society (table 3).5
The origin, development and regulation of LCH is complex and unravelling the molecular mechanisms that govern these processes are important for accurate diagnosis and treatment. LCH is characterized by a specific histopathological lesion containing clonal Langerhans-type cells, which can be recognized by their expression of CD1a, S100 protein, langerin (CD207) and Birbeck granules.6,7 Interestingly, based on their gene expression profile these pathological histiocytes do not correspond to epidermal Langerhans cells, but rather originate from myeloid-derived precursor dendritic cells.8 A 2010 report demonstrated recurrent genetic abnormalities in clonal LCH cells, establishing LCH as a neoplasm.9
To date, the two most prevalent mutations are found in either the proto-oncogenes BRAF (BRAFV600E) or MAP2K1.9,10 These mutations result in a constitutive activation of the mitogen-activated protein kinase (MAPK) pathway causing pathological proliferation and differentiation. However, not all responsible genes have yet been discovered (figure 1).11,12 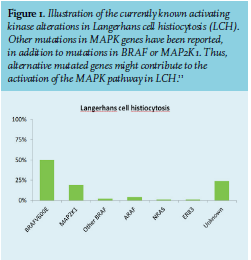
The endocrine system can also be affected by LCH, as Langerhans cells can migrate to the lymph nodes and affect the hypothalamic-pituitary axis. This can lead to mostly irreversible CDI with corresponding symptoms of polyuria and polydipsia. In Patient 1 and 3, CDI was the first presenting feature of LCH and in patient 2, CDI developed later in the course of the disease.
A positive water deprivation test can confirm the diagnosis of CDI. An MRI, typically shows a lack of the hyper-accentuated signal of the posterior pituitary on sagittal T1-weighted imaging and a thickening of the pituitary stalk.1 Infiltrative disease of the pituitary gland can lead to thyrotropic, somatotropic or gonadotropic axis failure.13 The prevalence of CDI in patients with LCH ranges from 10-50% and CDI presents in most cases after the diagnosis of LCH is established.1,13 In addition, CDI occurs more frequently in patients with BRAFV600E.14
Treatment options for LCH differ because of its wide disease spectrum. They range from minimal conservative treatment in single skin or bone disease to intensive combination chemotherapy in multisystem disease. However, vinblastine/prednisone treatment for one year is the standard initial systemic regimen for children.3
Adults, conversely, are preferably treated with cytarabine (second-line) because of poor overall responses and excessive toxicity of first-line chemotherapy.15 Targeted therapy with inhibitors of the BRAF/MAPK pathway (i.e. vemurafenib or trametinib, respectively) can be used in patients with corresponding mutations in whom first-line therapy fails; BRAFV600E is associated with more resistant LCH and higher reactivation rates in response to first-line treatment.14 LCH-induced CDI is treated with desmopressin.1 LCH prognosis depends on the extent of the disease, and disseminated disease may still have serious and even fatal outcomes.13
CONCLUSIONS
Our three patients clearly demonstrate that the diagnosis of LCH can be challenging and is sometimes made after a long clinical history. Occassionally, CDI arises as primary manifestation before LCH is known. Furthermore, although most publications report a male and pediatric predominance, our three cases presented females in their 40s and 50s. LCH is a rare disorder with a broad disease spectrum, and a more comprehensive understanding of the LCH etiopathogenesis may lead to alternative therapeutic interventions in the future.
ACKNOWLEDGMENTS
All authors declare no conflict of interest. No funding or financial support was received.
REFERENCES