KEYWORDS
Diagnosis, follow-up, iron, juvenile haemochromatosis, workup
INTRODUCTION
Hereditary haemochromatosis (HH) is a genetically heterogeneous disorder that results in body iron accumulation. The most common form is attributed to homozygosity for the p.Cys282Tyr (c.845G > A) variant of the haemochromatosis (HFE) gene on chromosome 6p21.1 Most male patients with HFE-associated hereditary haemochromatosis (HFE-HH; also called type 1 HH) do not present until middle age, and women not until after menopause; the disorder is mainly restricted to patients of Northern European descent.2
Other HH subtypes are rare and less restricted to certain populations.3-9 Juvenile forms of HH are caused by variants in genes encoding for haemojuvelin (HFE2 or HJV, located on chromosome 1q21, type 2A HH) and hepcidin (HAMP, located on chromosome 19q13, type 2B HH). These forms of HH are generally characterised by their early onset and a particularly severe phenotype, with patients typically presenting before the age of 30 with severe systemic iron overload, heart failure and endocrine disorders.10,11 Several studies have reported that of these two forms of juvenile haemochromatosis (JH), type 2A HH is by far the most common, comprising 90% of the approximately 110 reported patients diagnosed with JH.1,5,6,10-14 The phenotype of JH was first described in 1979 in a young woman with heart failure, insulin-dependent diabetes, amenorrhoea and hepatomegaly.14 In this report, 52 previously unreported cases were reviewed. In the following decades, the disease was further studied and outlined.15-17 In 2001, the JH phenotype was first linked to the HFE2 gene on chromosome 1 in a family of Greek descent.13-18
The haemojuvelin protein (HJV) is involved in the BMP receptor complex, which senses body iron stores and circulating iron, and subsequently stimulates hepcidin expression via the SMAD pathway.19 Pathogenic variants of HJV thus cause an inappropriately low level of hepcidin relative to body iron levels.20,21 This, in turn, leads to inadequate or ineffective hepcidin-mediated down-regulation of ferroportin on the basolateral membrane of enterocytes and the membrane of macrophages, and subsequent dietary increased iron absorption, relatively low iron content in the reticuloendothelial system and high parenchymal and circulating iron levels. Therefore, pathogenic variants of HFE2 lead to an HH phenotype (type 2A HH), and present for example with normal haemoglobin (Hb) levels, elevated ferritin levels and transferrin saturation (TSAT), and iron overload in the parenchymal tissues, such as the liver and exocrine pancreas.13
In order to define knowledge gaps and to assess the need for improvement of awareness and clinical management of type 2A HH in the Netherlands, we retrospectively collected genotype, biochemical and clinical presentation, diagnostic workup and treatment strategy in two women and five men (seven patients total; five probands and two siblings) from six families who were genetically and clinically diagnosed with type 2A HH in the Netherlands.
MATERIALS AND METHODS
Patients
We retrospectively collected and reviewed data on clinical presentation, biochemical tests and DNA analysis of seven patients who were genetically diagnosed with type 2A HH at the Radboud University Medical Centre between 2006 and 2016. This includes one patient who has been described previously,22,23 for whom we now provide follow-up data.
Laboratory methods
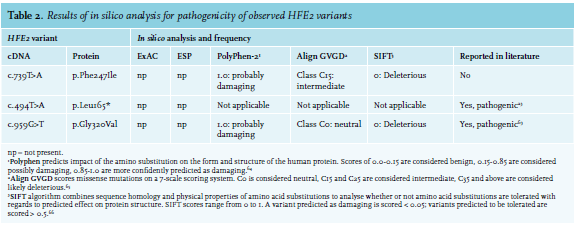
Liver iron content (LIC) was estimated by liver biopsy or magnetic resonance imaging (MRI) with a T1-weighted sequence. Free testosterone levels were calculated as described by Ross et al.24 Genotyping was performed by DNA sequence analysis of the full coding part of the genes and intron-exon boundaries, using Sanger sequencing. The pathogenicity of identified variants was assessed by association of the variant with the phenotype within a family, in silico tools and review of the literature. The in silico analysis was performed with Alamut Visual (Interactive Biosoftware), which comprises several predictive programmes to assess the consequence of missense and splice site mutations.25
CASE REPORTS
Patient 1
Patient 1 is a 62-year-old male of Dutch descent. At age 16, family screening was performed after his sister was diagnosed with HH, which revealed increased serum iron levels and heavy (grade IV) iron accumulation in hepatocytes.22 At that time, he complained of lower back pain and had freckled skin. His glucose tolerance test was normal. His TSAT was elevated (95%) and a Desferal test revealed a urine iron removal of 159 µmol iron/24 h (reference range (ref) < 35 µmol iron/24 h). Workup consisted of a liver biopsy, where LIC was found to be 324 µmol per gram dry weight (= 18 mg/g; ref < 36 µmol/g), in the presence of an intact liver architecture (table 1).22 Weekly phlebotomies were administered upon diagnosis of HH. During maintenance therapy, from ages 45 to 56, his ferritin values varied between 12 and 120 µg/l, and his TSAT between 13.4% and 87.0%. Alanine-aminotransferase (ALAT) was within the normal range.
At age 50, the patient was found to be homozygous for the HFE2 variant c.494T > A (p.Leu165*) (table 2).23 Despite a healthy lifestyle, he was diagnosed at age 52 with diabetes and hypertension and since then, has been taking metformin (table 1). At the age of 56, the patient developed a cerebrovascular accident, for which anticoagulant treatment was initiated. Further examination revealed a patent foramen ovale (PFO) and 50-90% stenosis of the carotid arteries. It is unclear whether his type 2A HH contributed to the development of his diabetes and cardiovascular symptoms. Currently, he still undergoes maintenance phlebotomy therapy 3-4 times per year.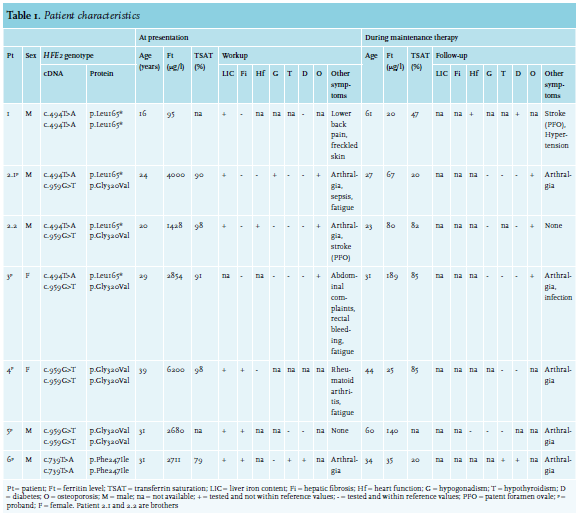
Patient 2.1
This patient is a 28-year-old male of Dutch descent. He presented at age 21 with ejaculatory failure and arthralgia of knees, elbows and hands. At the age of 23, he developed Yersinia enterocolitica sepsis and was subsequently hospitalised in the intensive care unit (ICU). During a check-up visit six months later, his ferritin level was severely elevated (4000 µg/l). The patient was clinically diagnosed with HH and started weekly phlebotomy treatments. Full evaluation showed a ferritin level of 2259 µg/l, with increased TSAT (99%) and immeasurably low testosterone level (< 0.1 nmol/l; ref 10.5 - 37 nmol/l), low luteinising hormone (LH) and follicle-stimulating hormone (FSH) levels (both < 1 U/l; ref in men 1.5-11 U/l and 1.4-8.5 U/l, respectively), indicating secondary hypogonadism, for which the patient started treatment with testosterone gel. The thyroid axis was unaffected. Shortly after, the patient developed an intercurrent E. coli sepsis as well. Genetic testing at this time showed compound heterozygosity for the HFE2 variants c.494T > A (p.Leu165*) and c.959G > T (p.Gly320Val) (table 2).
Workup in the two years after diagnosis consisted of several tests. Bone densitometry revealed osteopenia of the lumbar spine and femur collum. ALAT was found to be increased (90 U/l). An MRI scan of the liver and heart showed an LIC of > 350 µmol/g dry weight and cardiac iron content within reference values. Stroke and end-diastolic volume of the heart also remained within reference values. Testosterone was still low (1.6 nmol/l) with FSH and LH below the detection limit (< 1 U/l). The patient eventually switched from testosterone gel to subcutaneously applied choriongonadotrophin, which raised his testosterone level to 33.0 nmol/l and resolved his sexual problems.
As weekly phlebotomies did not appropriately decrease his ferritin level (3600 µg/l after six months of treatment), the patient switched to erythrocytapheresis (once per two weeks) at age 24, which lowered his ferritin value to 67 µg/l over the course of two years. Thereafter, maintenance therapy consisted of four phlebotomies per year.
After iron depletion, arthralgia decreased but did not completely disappear. Bone densitometry revealed bone density of the lumbar spine to be significantly improved, but no significant change was seen for the femur collum. Blood free thyroxine (FT4) levels remained within reference values (table 1). His latest ferritin level, at age 28, is 69 µg/l with an elevated TSAT of 85%.
Patient 2.2 This patient is 24 years old and the brother of Patient 2.1. He first presented with a cerebellar stroke at the age of 17. Diagnostic workup for this event resulted in identification of a PFO.
Upon clinical diagnosis of his brother, family screening at age 20 revealed an increased ferritin value of 1188 µg/l with an elevated TSAT of 90%. Furthermore, the patient reported mild arthralgia of the ankle, but no gonadal problems. His ALAT was normal. The patient was clinically diagnosed with HH and weekly phlebotomies were started. Initially, ferritin inexplicably rose to 2007 µg/l after one month, but after two-and-a-half months and eight phlebotomies, his ferritin level decreased to 1428 µg/l with a TSAT of 99% and kept decreasing during the following months.
Diagnostic workup took place during the two years post clinical diagnosis. Genetic screening confirmed compound heterozygosity for the HFE2 mutations c.494T > A (p.Leu165*) and c.959G > T (p.Gly320Val) (table 2). Bone densitometry revealed osteopenia of the lumbar spine. MRI of the liver revealed increased LIC of 310 (± 50) µmol/g dry weight, whereas iron levels in his heart were within reference values. Heart function was assessed as normal by ultrasound, and hormone analysis showed normal thyroid function. Four months into his phlebotomy treatment, at a ferritin level of 1125 µg/l, testosterone was low (3.1 nmol/l); FSH and LH were not measured at this point in time. Several months later, the testosterone (10.6 nmol/l) and free testosterone (120 pmol/l; ref 120-750 pmol/l)26 were just within reference range. FSH and LH were normal as well. No other abnormalities were reported (table 1). One year after the first testosterone measurement, testosterone levels had increased to 20.5 nmol/l (free testosterone 266 pmol/l) with a ferritin value of 147 µg/l. At 24 years of age, TSAT was 82% and depletion of iron stores was achieved with a ferritin level of 80 µg/l (table 1). Thereafter, maintenance phlebotomy therapy consisting of six phlebotomies per year was started. Upon iron depletion, bone density of the lumbar spine, but not that of the femur collum, was found to be significantly improved, and the patient did not report arthralgia.
Patient 3
This patient is a 32-year-old female proband of Dutch descent. She had had complaints of severe fatigue and several infections since childhood, but despite extensive hospital examination, the cause was not identified until age 29. At this age, she presented with abdominal complaints and rectal bleeding. Evaluation revealed an elevated TSAT (91%) and a severely increased ferritin value (2854 µg/l). Although the common HFE variants (C282Y and H63D) were not identified, the patient was diagnosed with HH and weekly phlebotomies were planned. Almost a year after starting treatment, having received only 13 phlebotomies due to lack of compliance, her ferritin level was 1994 µg/l, and she presented with arthralgia in her wrists and fingers. Genetic testing revealed compound heterozygosity for c.494T > A (p.Leu165*) and c.959G > T (p.Gly320Val) in the HFE2 gene (table 2).
During the two years after diagnosis (age 29-31 years), workup consisted of i) bone densitometry revealing osteoporosis of the lumbar spine and femur collum; ii) computed tomography scan of the hand revealing diffuse osteopenic aspect; iii) analysis of TSH, which was found to be within the reference range; and iv) evaluation of the gonadal axis, which showed no abnormalities (table 1). ALAT was normal and ultrasound of the abdomen showed no indication for the presence of cirrhosis or hepatocellular carcinoma. Further examinations to assess liver iron and imaging studies of the heart were not performed.
At age 30, at a ferritin level of 838 µg/l, the patient switched to biweekly phlebotomies, since she found the weekly phlebotomies too taxing. At this age, a Pseudomonas infection of the peripherally inserted central catheter (PICC) was found. At age 31, the TSH was still within reference range and her ferritin levels reached 189 µg/l (table 1). Currently, the patient is doing well, with enough energy to work full time.
Patient 4
This patient is a 44-year-old female proband of Dutch descent. At age 39, she presented with seronegative rheumatoid arthritis of the metatarsal-phalangeal and proximal interphalangeal joints and reported fatigue. Laboratory testing showed a severely increased ferritin level of 6200 µg/l with elevated TSAT (98%) and ALAT (150 U/l) levels. Weekly phlebotomies were started upon clinical diagnosis of HH, after which arthralgia persisted. In the workup of the following months, no bone densitometry was performed. Echography and subsequent MRI of the liver revealed multiple lesions (not metastases) as well as steatosis hepatis, iron accumulation and mild fibrosis, but no hepatomegaly. Using tissue Doppler imaging, which may be used as a screening tool to detect cardiac involvement in patients with HH, no cardiac iron accumulation was found. Liver biopsy revealed severe (grade IV) iron accumulation (table 1). TSH, FT4 and the gonadal axis were not evaluated at this point in time. After eight months of phlebotomy, her liver enzyme levels had normalised. Three years after presentation, her TSH was high normal (4.2 mU/l), FT4 was normal and diabetes mellitus was absent. Ferritin levels varied between 24 and 64 µg/l with a TSAT around 85%. Four years after clinical diagnosis, genetic testing revealed a homozygous c.959G > T (p.Gly320Val) variant in HFE2 (table 2). Her arthralgia has decreased, but not completely disappeared (table 1).
Patient 5
This patient is a 60-year-old male proband of Dutch descent. He was first noticed in routine medical evaluation at age 31, as his liver enzymes were elevated with a ferritin of 2680 µg/l. Diagnostic workup consisted of liver biopsy showing grade IV iron accumulation with fibrosis. Blood tests revealed normal thyroid status: TSH and total thyroxine were within normal limits (table 1). HH was diagnosed and phlebotomies were started. Evaluation of the gonadal axis and bone density was considered but not deemed necessary. At age 55, the patient presented with arthrosis in both halluces (table 1). Of note, this had existed for 11 years, during which arthrodesis had been performed. The patient’s treatment at the time consisted of phlebotomies once every eight weeks. Ferritin was 140 µg/l. Genetic testing of the HFE2 gene revealed homozygosity for the pathogenic variant c.959G > T (p.Gly320Val) (table 2). After this diagnosis, an ECG and ultrasound of the heart were performed, and no abnormalities were found. Iron loading of the heart was not tested. Laboratory results did not indicate any abnormalities of liver, gonadal axis or thyroid. His most recent ferritin level, measured at age 60, was 29 µg/l. He still undergoes phlebotomies once every eight weeks and feels well, although he does have painful shoulders and wrists.
Patient 6
This patient is a male proband, 35 years of age and of Turkish origin. At age 31, he presented with arthralgia in his hands, fatigue and weight loss and was diagnosed with diabetes and microcytic anaemia (Hb 7.8 mmol/l, mean corpuscular volume 78 fl). Diagnostic workup of his anaemia showed heterozygous β-thalassaemia (or β-thalassaemia minor), a TSAT of 79% and a ferritin value of 2711 µg/l. Furthermore, MRI of the liver yielded LIC in excess of 350 µmol/g dry weight and a liver biopsy identified severe iron accumulation in the hepatocytes and mild fibrosis. T2* imaging of his heart showed normal cardiac iron content. ALAT was high normal (54 U/l). His testosterone, LH and FSH levels were within reference ranges. The patient also had subclinical hypothyroidism: TSH levels were increased (7.6 mU/l, ref 0.4-4.0 mU/l) and FT4 was normal (table 1). Combined, and since β-thalassaemia minor is not associated with iron overload, these findings led to the clinical diagnosis of HH, and weekly phlebotomies were started.
At age 31, genetic diagnosis showed that the patient was a homozygous carrier of the c.739T > A (p.Phe247Ile) HFE2 variant. This variant was not previously described in literature, but was predicted as pathogenic by in silico analysis (Align GVGD, SIFT, PolyPhen-2) (table 2). His brother and sister were diagnosed with the same HJV genotype and both show a phenotype consistent with type 2A HH. However, as they never presented at our centre, we do not have any clinical data on these two siblings.
Phlebotomies were well tolerated and at age 34, iron depletion was achieved (TSAT 20%; ferritin 35 µg/l) and maintenance therapy with phlebotomies every three months was planned. However, due to lack of compliance, the patient did not receive therapy for one year, during which TSAT and ferritin levels increased to 76% and 135 µg/l, respectively. Currently, his diabetes is well regulated with metformin and his hypothyroidism has remained subclinical with a TSH of 6.1 mU/l and an FT4 of 14 pmol/l (ref 8.0-22.0 pmol/l). His ALAT has normalised, but a mild arthralgia is still present.
DISCUSSION
In our sample consisting of seven patients from six families diagnosed with type 2A HH in the Netherlands, we observed: i) a highly pleiotropic presentation; ii) a significant delay to the clinical diagnosis in two patients; iii) only two variants that are responsible for the six biallelically-affected patients from five families of Dutch origin; iv) a practice variation in the diagnostic workup, follow-up and applied treatment strategy; and v) novel complications that may be attributed to type 2A HH occurring with ageing despite the long-term iron depletion therapy.
Probands (three men and two women) first presented in adulthood, but their ages varied. Female probands presented at a later age (range 31-39) than male probands (range 19-31). Our female patients had not been pregnant prior to presentation, but blood loss (and thus loss of iron) due to menstruation may explain the later onset. Findings of a JH case series and a study comparing JH with type 1 and 3 HH are in agreement with our findings.10,11 The prevalence of presenting symptoms of our seven patients compared to three previously reported case series of JH patients (N, respectively 13 (> 8 years old, defined as < 30 years old at presentation), 26 (adults, defined as presentation < 30 years old), 37 (adults, linked to chromosome 1q)) can be summarised as follows: arthralgia 57% in our patients vs. 27-30% in previous reports; hypogonadotropic hypogonadism 14% vs. 77-96%; diabetes/reduced glucose tolerance 29% vs. 31-58%; and cardiac problems 29% vs. 35-54%.10-12 In addition, we report one patient out of seven with subclinical hypothyroidism, in comparison to one out of 13 that had hypothyroidism in one of the aforementioned case series.12 The others do not report hypothyroidism, which corresponds to the notion that the development of hypothyroidism in juvenile haemochromatosis is rare.12,28,29 Hashimoto thyroiditis characterised by thyromegaly, hypothyroidism and elevated serum concentrations of anti-thyroid peroxidase (anti-TPO) antibodies has been documented as a cause of thyroid dysfunction in two patients suspected for JH.30 To the best of our knowledge, it is not fully known whether hypothyroidism in JH is coincidental, a primary thyroid dysfunction or secondary anterior pituitary failure.29,30
Three of our patients were diagnosed with osteopenia or osteoporosis, one of whom also had hypogonadism. Osteopenia and osteoporosis are reported as common complications in type 2A HH.31 Proposed underlying mechanisms include hypogonadism, liver failure and iron overload,32 of which liver failure is unlikely since liver fibrosis was absent in our patients diagnosed with osteoporosis. Furthermore, one patient developed sepsis from Yersinia enterocolitica and E. coli in his early 20s. From the literature, these infections have been described in states of iron overload and related high iron availability, and are the result of hepcidin deficiency. Among the causal micro-organisms are – apart from Yersinia enterocolitica and E. coli – also Vibrio vulnificus and Listeria monocytogenes. 33-38 Another patient (patient 3) presented with a central-line infection caused by Pseudomonas, which has also been associated with iron overload.39
Overall, the differences in prevalence of most of the presenting symptoms in our series compared to the literature might be characterised as chance findings due to our small sample size. The lower prevalence of most symptoms may be attributed to rising timely diagnosis and treatment of patients in the last decade, as recently reviewed.6
Two out of our five probands (patients 2.1 and 3) had a significant delay to clinical diagnosis. One of these probands, patient 2.1, presented with multiple concomitant symptoms: arthralgia, hypogonadism and sepsis. Delay to clinical diagnosis has been reported by others in patients with type 2A HH,40 who developed severe cardiac complications. Since severe complications also occurred in one proband (patient 2.1) with delay to clinical diagnosis, we urge physicians to be mindful of possible iron overload if systemic symptoms in young patients go unexplained for several months.
In this respect, for patients without HFE C282Y homozygosity or C282Y/H63D compound heterozygosity and with hyperferritinaemia and TSAT > 45%, existing guidelines41 recommend direct assessment of liver iron by MRI or liver biopsy. If iron excess has been proven (i.e. > 3-6 times the upper limit of normal)42,43 and other (hepatic or haematological) diseases have been ruled out, genetic testing for rare defects in HFE and other non-HFE haemochromatosis genes should be performed.
We found the previously unreported c.739T > A (p.Phe247Ile) variant in our non-Dutch patient. Moreover, we report that the common c. 959C > G (p.Gly320Val) variant and the c.494T > A (p.Leu165*) variant to be the causative mutations for all of our patients of Dutch descent. To date, the latter variant has only been described in Dutch patients,23 suggesting a founder effect. Since numbers are low, we were unable to assess if there was a difference in iron accumulation between patients with the different genotypes. To the best of our knowledge, a genotypephenotype relationship between HFE2 mutations and iron accumulation has not yet been described in the literature. Alternatively, other genes or factors aside from menstrual blood loss may play a modifying role in rate of body iron accumulation.
We found that after the diagnosis of type 2A HH was made, workup, follow-up and treatment strategies differed between subjects. Currently, no guideline or evidence exists for the optimal workup, follow-up and treatment of type 2A HH. Nevertheless, workup for organ damage by iron accumulation is generally performed by physicians in JH patients, prompted either by genetic diagnosis or abnormally elevated ferritin levels. In the current case series, this workup was variable: heart function and the gonadal axis were only studied in some of the patients, and evaluation of liver iron content by MRI (n = 2), liver biopsy (n = 2) or both (n = 2) was performed in all subjects except one. For the latter patient, ultrasound of the liver was performed, but this technique is not suitable for the detection of iron overload. Of the four patients who presented elevated liver enzymes at diagnosis, a liver biopsy was performed in three of them. Follow-up also differed between patients, with symptoms of hypogonadism and glucose intolerance-initiated workup in two patients. However, in the absence of complaints, the patients were not always tested for endocrine abnormalities and possible associated osteoporosis.
In the absence of guidelines for treatment of type 2A HH, treatment strategies for the patients described here were similar to that of HFE-HH,6 as laid out in the European Association for the Study of the Liver (EASL) Guidelines for HFE haemochromatosis.41 In accordance with the EASL Guidelines, six out of seven patients were phlebotomised until iron depletion was achieved; of these, five underwent weekly phlebotomies to obtain full depletion. One patient however, switched to biweekly phlebotomies and another switched from phlebotomy to erythrocytapheresis. In all patients, this depletion therapy took at least 24 months. Notably, we observed variation between patients in target ferritin levels for both the depletion and maintenance phase. Erythrocytapheresis was successfully used for iron depletion in one of the patients. A recent observational study showed beneficial effects of this therapy.44 However, a Cochrane review concluded that there is currently insufficient evidence to determine whether erythrocytapheresis is beneficial or harmful compared to phlebotomy therapy.45
While iron-depleted, two of our patients developed new (co)morbidities with ageing. These comprised stroke, diabetes and articular problems. Although the prevalence of these morbidities increases with ageing in the general population,46 it is also conceivable that they can be attributed to HJV function in cells other than hepatocytes, since HFE2 is also expressed in myocardial cells,13,47 and β and acinar cells of the pancreas.48 Another possibility is that long-term exposure to non-transferrin bound iron (NTBI) contributes to the development of these morbidities. Indeed, even after iron depletion, in the maintenance phase, TSAT in most patients remains elevated (above 70%), producing NTBI in the circulation.49 NTBI has been documented as toxic for parenchymal cells,47,50-54 including pancreas β cells and cardiomyocytes, where it has been reported to be taken up in an unregulated manner by ZIP14 or L-type Ca2+ channels.55,56 Interestingly, increased circulating iron has been associated with increased atherosclerosis in a mouse model of hereditary haemochromatosis57,58 and with coronary artery disease in patients with stable angina pectoris.59 Long-term exposure to high TSAT-induced NTBI levels may also predispose patients to arthralgia. Indeed, articular manifestation is a common occurrence in type 2A HH,10-12,31 but how exactly iron overload damages joints in HH – in general and in JH in particular – is not yet clear.60,61 Therefore, we speculate that the chronic presence of NTBI underlies the development of complications in type 2A HH over time, despite the depletion of patient iron stores. If this is indeed the case, adequate treatment remains a challenge: phlebotomy therapy to lower TSAT in these patients would require extensive venesection, which could lead to anaemia. It is however, conceivable that novel hepcidin agonists, which are currently under development as novel therapies for diseases that are characterised by low hepcidin levels relative to body iron stores,62 may provide a solution in the future.
In conclusion, even though we note that awareness of type 2A HH is increasing among physicians, much remains unclear for the optimal management of this disease regarding workup and follow-up, and for the significance of chronically elevated TSAT for the development of long-term complications in patients with type 2A HH. Therefore, we advocate research on the natural course of the disease and the cost-effectiveness of the various workups, follow-ups and diagnostic strategies. Timely referral, a global registry and management of these patients within networks of expertise centres, such as the recently initiated European Reference Networks, will help achieve these goals. Until this has been accomplished, we recommend to adhere to existing clinical guidelines for HFE-HH: to screen for the HFE2 genotype and iron overload phenotype of all first-degree relatives, and to assess diagnosed patients for complications including liver fibrosis and cirrhosis, diabetes mellitus, joint disease, endocrine deficiency (hypothyroidism and hypogonadism), cardiac disease and osteoporosis, preferably before the start of intensive iron depletion therapy.
ACKNOWLEDGEMENTS
We would like to thank P.W. Friederich for his help in collecting the data and J.P.H. Drenth for his critical revision of the article.
DISCLOSURES
All authors declare no conflict of interest. No funding or financial support was received.
REFERENCES