
KEYWORDS
IgG4-related disease (IgG4-RD)
INTRODUCTION
IgG4-related disease (IgG4-RD) is a rare immune-mediated inflammatory disorder that predominantly affects elderly men (over 65 years of age). Calculated incidence rates in Japan are 2.8-10.8 per 1 million people.1 It is characterised by tumefied lesions that can affect various organs simultaneously or consecutively.
IgG4-RD was not recognised as a systemic condition until the early 2000s when extra pancreatic manifestations were identified in patients with autoimmune pancreatitis type 1.2 Many medical conditions that have long been viewed as conditions confined to single organs (e.g. Küttner’s tumour, Mikulicz’s disease, retroperitoneal fibrosis, the fibrosing form of Hashimoto’s and Riedel’s thyroiditis) are now considered to be part of the spectrum of IgG4-RD. Although IgG4-RD is increasingly gaining international attention, it is still an unrecognised disease, even more so in children. A recently published article provides an overview of all reports published on IgG4-RD in children.3 We present two patients with IgG4-RD, who had symptoms for many years. Prior investigations, including tissue biopsies, failed to diagnose the disease.
CASE REPORTS
Patient 1
A 72-year-old man was examined because of a renal mass in his right kidney and pulmonary nodules suggestive of malignancy (figure 1). Medical history revealed prostate cancer (2012), abdominal aortic aneurysm and retroperitoneal fibrosis (2006) with bilateral hydronephrosis that resulted in functional loss of the right kidney. A nephrostomy catheter was inserted for the left kidney.
In 2014, a PET-CT scan showed several fluorodeoxyglucose (FDG) positive pulmonary nodules and FDG uptake in the thyroid gland for which a diagnostic hemithyroidectomy was performed after cytology remained inconclusive. Histology showed diffuse inflammation. Diagnosis of Hashimoto’s thyroiditis was made based on radiological and biochemical characteristics.
The patient complained of fatigue and dyspnoea. Several biopsies of the pulmonary lesions showed chronic inflammation and fibrosis, but no malignancy. Because high suspicion of malignancy remained, a wedge excision of the lung was performed. Histology showed significantly increased plasma cell infiltration and focal storiform fibrosis but no obliterative phlebitis (figure 2). Additional immunostaining showed 62/high-power field (HPF) IgG4-positive plasma cells and an IgG4/IgG ratio of 0.42 (figure 3). Serum IgG4 was elevated. Based on these results, IgG4-RD was diagnosed.
Revision of previously performed biopsies (with additional IgG4 immunostaining) also showed findings characteristic of IgG4-RD in the retroperitoneum and thyroid (e.g. the IgG4-related form of Hashimoto’s thyroiditis), but not in the prostate.8 The aortic aneurysm was assumed to have been caused by IgG4-related aortitis.
A repeated PET-CT scan showed increased FDG uptake in the pulmonary nodules compared with the PET-CT performed in 2014. The renal mass turned out to be a cyst. Treatment with 60 mg prednisone was initiated for two weeks. The dose was tapered to 45 mg after four weeks. A new PET-CT scan showed decreased FDG uptake in the pulmonary nodules. The serum IgG4 level declined and the dose was further tapered (table 1).
The patient remained clinically stable until he unexpectedly died of gastrointestinal bleeding without a clear cause, six months after diagnosis. A link with IgG4-RD or prednisone was not considered likely but could not be excluded because no autopsy was performed. 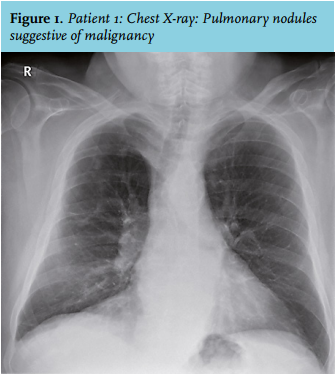
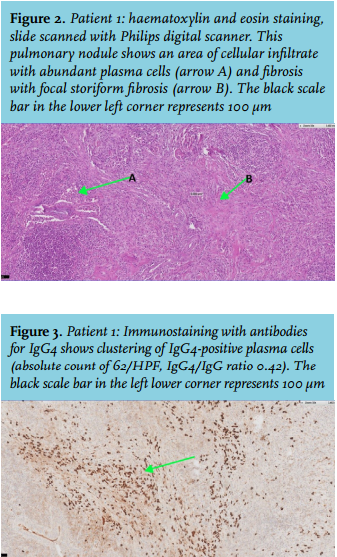
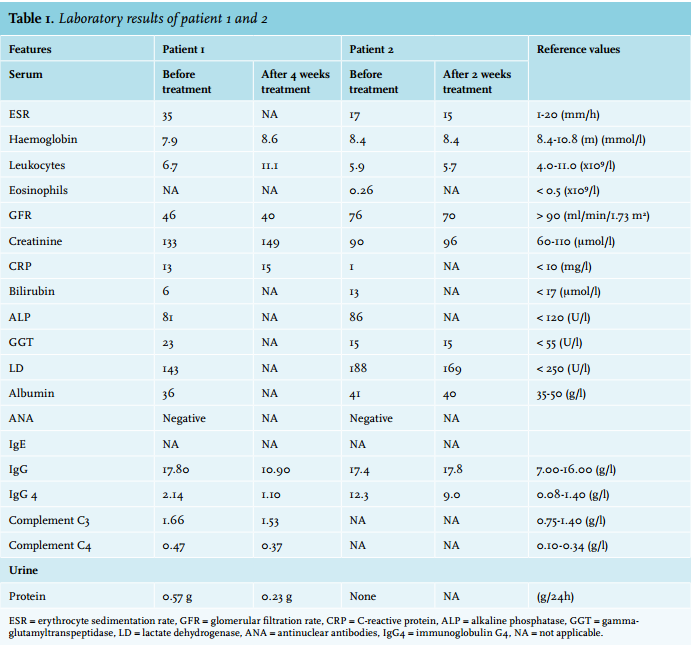
Patient 2
A 56-year-old man was examined for painless cervical and submandibular lymphadenopathy that had been progressive for two weeks. His other complaints were fatigue and generalised new-onset pruritus. He had had chronic cervical lymphadenopathy for several years and earlier investigations failed to diagnose its cause. A lymph node biopsy in 2010 showed lymphoid cells and a starry sky pattern of macrophages. Medical history revealed hypothyroidism and erythema migrans, both of which were treated. A PET-CT scan showed FDG-positive cervical, mediastinal and hilar lymphadenopathy, and two (7 mm) intrapulmonary lesions. There was no FDG uptake in the thyroid. Lymph node biopsy showed a similar pattern as was seen in 2010. Reanalysis for IgG4-RD was requested on the biopsy taken in 2010 and showed 82/HPF IgG4 positive plasma cells and a IgG4/IgG ratio of 0.66 but without fibrosis or phlebitis. Serum IgG level was elevated (table 1). Dermal biopsy reported no positive IgG4 plasma cells.
IgG4-RD with lymphadenopathy and possible IgG4-related hypothyroidism and dermatological involvement (though not biopsy proved) was diagnosed. Prednisone 40 mg was started. Evaluation after two weeks showed clinical response with a decrease in lymph node size of 50%. The serum IgG4 level declined and a positive effect on his dermatological symptoms was reported. Prednisone was tapered over months without signs of recurrence during prednisone withdrawal.
DISCUSSION
Here we present two patients in whom IgG4-RD had been present for several years but was not recognised despite prior tissue analyses. Revision of these biopsies for specific features of IgG4-RD proved to be very valuable in the diagnostic process.
Awareness of IgG4-related disease is important as it can affect many organs, although it has certain sites of preference such as the pancreas, orbit, salivary tract and lymph nodes. Clinical symptoms vary according to the spectrum of organs involved. IgG4-RD has been identified to mimic several other diseases such as infections and malignancies and the differential diagnosis is broad.5,7
A great diversity of clinical and radiological presentations of pulmonary IgG4-RD has been described and diagnosis can be very difficult.6,7
In patient 1 extensive pulmonary analysis excluded malignancy. Features of IgG4-RD were present in the lung and prior biopsies of retroperitoneum and thyroid. In patient 2 mainly lymph nodes were involved, which is a more common presentation of IgG4-RD. Thyroid, pulmonary and skin involvement was suspected but not proven by biopsy.
Both of our patients had elevated serum IgG4 levels at baseline. Though initially viewed as a major diagnostic biomarker, it soon became clear that serum IgG4 lacks adequate sensitivity and specificity for diagnostic purposes when used as a solitary marker.9,10 However, serum IgG4 levels may be of assistance in the diagnostic process in some cases.9 Inflammatory markers are not specific for IgG4-RD.
Definite diagnosis of IgG4-RD requires both histopathological information and clinicopathological correlation. Histopathology remains the cornerstone in diagnosis with dense lymphoplasmacytic infiltrates, rich in IgG4-positive plasma cells, storiform fibrosis and obliterative phlebitis as key features. In our both patients the Boston consensus criteria for IgG4-RD were met.8 A FDG-PET scan (as performed in both patients) is very useful to determine organ involvement and monitor disease activity.11
IgG4-RD is thought to be rare, but is probably often overlooked. In both patients it took several years before the diagnosis was made. Prior investigations, including tissue biopsies, failed to diagnose the disease. At that time, the treating clinicians were not aware of the possibility of IgG4-RD and immunostaining for IgG4-positive plasma cells is not a standardly performed procedure in our histopathological analysis.
Attention towards IgG4-RD in pulmonary pathology triggered the pathologist to perform additional immunostaining for IgG4-positive plasma cells when a markedly raised number of plasma cells was seen in the wedge biopsy in patient 1. These results in combination with the patient’s symptoms and medical history led to the diagnosis and revision of the previous biopsies. In patient 2 we were more alert to the possibility of IgG4-RD since we had recently diagnosed IgG4-RD in patient 1. We specifically asked for a revision of the biopsy for specific IgG4-RD features, which led to diagnosis.
The pathogenesis of IgG4-RD, and more specifically the role of the IgG4 (auto)antibodies, is still not entirely clear and is the subject of research. In general theory, an antigen-driven immune response drives B cells/Th2 cell to IgG4 production and fibrotic alternations by inflammatory cytokines, such as interleukin (IL)-4, -5, -10, -13 and transforming growth factor (TGF)-β. This inflammation and fibrosis leads to tissue infiltration and swelling.12
IgG4-RD often follows a subacute course with vague non-specific symptoms and spontaneous regression may occur. Remission, however, is often temporary and most patients require therapy in order to prevent irreversible organ damage.13 Once fibrosis is established, therapeutic options are limited. At the time of diagnosis irreversible organ damage was already present in patient 1 (e.g. functional kidney loss due to retroperitoneal fibrosis, aortic aneurysm and hypothyroidism). In both patients treatment was initiated in order to relieve symptoms and prevent future organ damage.
According to the recently published international consensus guidance statement, steroids (initial dose 40-60 mg prednisone) are the first treatment choice and steroid-sparing agents such as azathioprine may be considered as an add-on therapy for patients with a relapse under steroid treatment.13 Experts are divided on the question whether steroid-sparing agents should be used in combination with glucocorticoids from the start of treatment.13 Recent articles show promising results of rituximab as a treatment for IgG4-RD.14,15
The definite role for steroid-sparing agents and rituximab has to be further established. Recently, good predictors of relapse have been determined in patients treated with rituximab. The higher the baseline values of serum IgG4, IgE, and eosinophils, the greater the risk of relapse and the shorter the time to relapse.15 Both of our patients responded well to steroids with an improvement in their clinical symptoms, a decrease in IgG4 serum levels and also radiological improvement in patient 1. A PET-CT scan will soon be repeated in patient 2. Patient 1 did not show any signs of a relapse while still on a low dose of steroids at the time he unexpectedly died of gastrointestinal bleeding. Patient 2 remained clinically stable after steroid withdrawal. In case of a relapse, treatment with steroid-sparing agents will be considered.
In conclusion, awareness of IgG4-related disease is important, as it has been shown to mimic other diseases. Delay in treatment may result in irreversible organ damage. We emphasise that, if IgG4-RD is suspected, a specific tissue analysis for IgG4-RD features (with immunostaining for IgG4-positive plasma cells) is requested. In this light, revision of old biopsies may be necessary.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES