KEYWORDS
Fibro-inflammatory disease, immunoglobulin G4, and IgG4-related disease
INTRODUCTION
Immunoglobulin G4-related disease (IgG4-RD) is a systemic fibro-inflammatory condition with manifestations in almost all parts of the human body.1 It is characterised by tumour-like infiltration of IgG4-positive plasma cells in the tissues, mostly with fibrotic or sclerotic abnormalities, and often elevated serum IgG4 levels.1 IgG4-RD was initially described in patients with sclerosing pancreatitis, but from 2003 it is recognised as a systemic disease.2 The disease can manifest in one single organ, but it can also occur simultaneously in multiple organs. IgG4-RD usually occurs in the salivary and lacrimal glands, the orbit, pancreas and lymph nodes. Other preferential localisations include the lungs, kidneys, thyroid, peritoneum and prostate.3 Conditions previously called Mikulicz’s disease, sclerosing sialadenitis, inflammatory orbital pseudotumour, a subset of idiopathic retroperitoneal fibrosis and Riedel’s thyroiditis are now reclassified under the umbrella of IgG4-RD.4 IgG4-RD mimics many infectious, inflammatory and malignant disorders often leading to a delay in both diagnosis and treatment, potentially progressing into irreversible fibrosis.5 Awareness of this disease is important to avoid unnecessary delay. We therefore present three different cases of patients with IgG4-RD to emphasise the broad clinical presentation of this disease and present a review on the pathogenesis, diagnosis and treatment.
CASE PRESENTATIONS
We briefly present three different cases of IgG4-RD. The patient characteristics and the main clinical features are shown in table 1.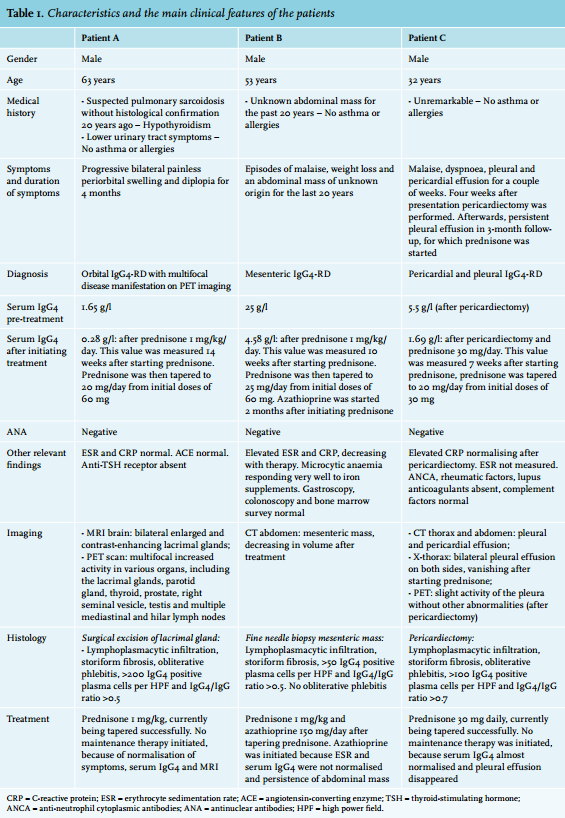
Patient A
This 63-year-old male patient was referred to the ophthalmologist because of a painless bilateral periorbital swelling and diplopia suspected to be lymphoma or recurrence of sarcoidosis. Pulmonary sarcoidosis was diagnosed on the basis of the clinical symptoms and was not histologically confirmed, and this had been stable without medication for 20 years. His history also included levothyroxine for hypothyroidism and alpha-blockers for relapsing lower urinary tract symptoms. Bilateral periorbital swelling with slight proptosis was found on physical examination. Laboratory tests revealed elevated serum IgG4 without other abnormalities. Computed tomography (CT) of the thorax and abdomen was normal. MRI of the brain revealed only bilateral enlarged and contrast-enhancing lacrimal glands (figure 1A). On F-18 FDG PET/CT scan, multifocal increased activity was noted in various organs (table 1). Histology of the lacrimal gland was compatible with IgG4-RD (figure 1C+D). Prednisone 1 mg/kg/day significantly decreased the periorbital swelling, but also resulted in a complete recovery of the urinary tract symptoms within one week and recovery of thyroid dysfunction. After four weeks, the steroids could be tapered and levothyroxine was discontinued without recurrence after six months of follow-up.
Patient B
A 53-year-old male patient consulted several medical specialists over the past 20 years because of an abdominal mass. Extensive diagnostics including biopsies and bone marrow examination did not yield any diagnosis. The patient complained of slowly progressive malaise, weight loss and abdominal pain. After referral to our hospital, IgG4-RD was suspected, also because of the elevated serum IgG4 levels. Laboratory tests further revealed an elevated erythrocyte sedimentation rate (ESR), normal ferritin and microcytic anaemia, known to have existed for years. Gastroscopy and colonoscopy were without evidence of malignancy, intraepithelial lymphocytosis, IgG4-RD, villous atrophy, Giardia, Whipple’s disease or Helicobacter pylori infection. CT imaging demonstrated a progressively increasing mesenteric mass of 50 mm surrounded by mesenteric lymphadenopathy (figure 2A). Histology of the mesenteric mass confirmed the diagnosis of IgG4-RD (figure 2C). Prednisone 1 mg/kg/day was initiated. Hereafter, the symptoms eased, the serum IgG4 and ESR decreased and haemoglobin levels almost normalised. The abdominal mass and lymphadenopathy decreased (figure 2B) and serum IgG4 and ESR levels showed a downward trend. The steroids were tapered after four weeks and azathioprine 150 mg daily was started after two months since the mass had not totally regressed.
Patient C
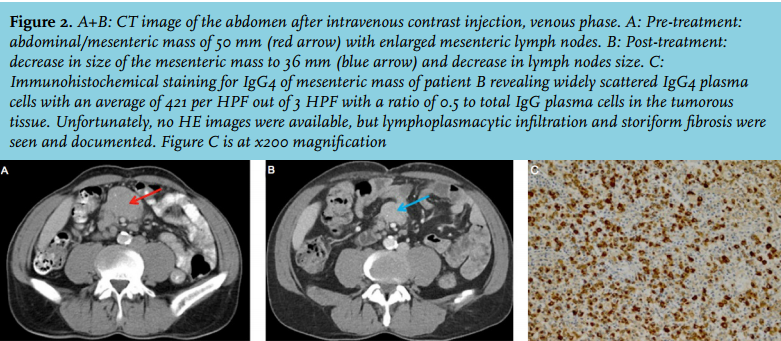
This 32-year-old male patient was admitted to the department of cardiology because of cardiac tamponade. On a CT of the thorax and abdomen both pleural and pericardial effusions were seen (figure 3A). Laboratory tests showed elevated C-reactive protein (CRP), ESR was not measured at that moment. Because of persistent pericardial effusion with constrictive signs, a pericardiectomy was performed and diuretics were given. Hereafter, the CRP normalised and the ESR was normal. Detailed bacteriological and virological analyses (including serology or viral load determinations of HIV, hepatitis A/B/C, Borrelia burgdorferi, syphilis, mycoplasma, tuberculosis, parvovirus, Cytomegalovirus, Epstein-Barr, Coxiella burnetii, toxoplasmosis, Coxsackie virus and varicella- zoster) were unremarkable. Elevated serum IgG4 and pericardial histology finally offered sufficient evidence for IgG4-RD (figure 3D+E). Cultures of the pericardial tissue ruled out bacterial pathogens including Mycobacterium tuberculosis. F-18 FDG PET/CT three months after pericardiectomy revealed slight activity of the pleura without other abnormalities. Prednisone 30 mg daily led to the disappearance of the pleural effusion (figure 3B+C) and almost normalisation of serum IgG4; diuretics were no longer required. Hereafter, the prednisone carefully was tapered to 20 mg in seven weeks without signs of recurrence.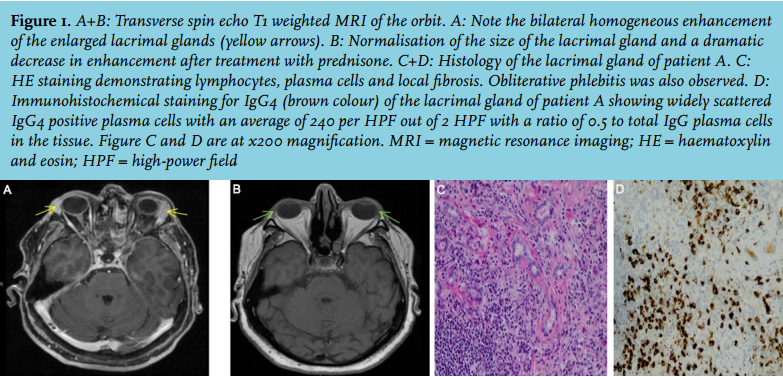
DISCUSSION
Here we present three cases of patients with unrecognised IgG4-RD, each presenting with a different clinical presentation. The courses of these patients reflect the broad spectrum of clinical faces of IgG4-RD. By demonstrating the variable presentation of Ig4-RD, we briefly provide an overview of the spectrum of symptoms and treatment options in this new disease entity.
IgG4-RD is a systemic disease that can be found in almost any organ, but with certain sites of preference (orbit, salivary tract, pancreas and lymph nodes) which may be guiding when considering this new disease entity. On the other hand, IgG4-RD mimics various benign and malignant disorders. Therefore, careful diagnostics should be applied before setting the diagnosis.1 The vast clinical manifestation range and potentially organ- and life-threatening situations emphasise that awareness of this relatively new entity is pivotal to swiftly set a diagnosis and prevent organ damage.3 This is highlighted by the histories of the presented patients. Patient A presented with a relatively short history, and lymphoma or recurrent sarcoidosis were suspected. Extensive diagnostics were conducted to rule out these entities. A typical FDG-uptake pattern led to the diagnosis of IgG4-RD by histology of a lacrimal gland. The abdominal mass resembling retroperitoneal fibrosis seen in patient B is remarkable and has rarely been described before.6 Multiple diagnostics including biopsies of the abdominal mass excluded conditions such as malignancy and infectious diseases. Eventually, after almost 20 years, attention towards IgG4 resulted in the diagnosis of IgG4-RD. Cardiac manifestations of IgG4-RD, such as in patient C, are rare.7 The patient presented with constrictive pericarditis and a pleural effusion. It remains a challenge to rule out infectious or malignant disease and consider IgG4-RD.
The diagnosis of IgG4-RD is based on the combination of clinical presentation, serological and histological findings, but histology is the gold standard. Although the disease is called IgG4-RD, about 30 to 50% of histologically proven cases show normal IgG4 levels leading to misinterpretation and erroneous rejection of the diagnosis.7 Furthermore, the specificity and positive predictive value of serum IgG4 concentrations are low, which make them poor disease markers. In our cases, serum IgG4 levels were elevated in all three patients, but with different ranges (1.65 to 25 g/l). Other, though unspecific, serological findings are ESR and CRP in patients with active disease, but these are elevated in 53% and 40%, respectively, of the cases.7 In this study 51% of these patients had elevated serum IgG4.7 In our patients, not all elevated IgG4 levels corresponded with an elevated ESR and CRP.
Only in patient A were the ESR and CRP both normal. Although speculative, longstanding active disease and high serum IgG could lead to elevated ESR and CRP, which applied in case B.
Measuring plasmablasts originating from CD20+ B cells is a superior alternative to measuring IgG4 concentrations in serum,8 but so far this technique has not been widely introduced for clinical application. So far, imaging studies play a crucial role in the diagnostics of IgG4-RD; however, imaging is not specific for this disease and several conditions such as malignancy should be excluded. Radionuclide imaging in patient A was more sensitive than conventional CT. Several studies have shown the usefulness of FDG-PET/CT scanning for diagnosis, staging and the degree of organ involvement and monitoring of therapy response, and this imaging method seems to detect more lesions than conventional methods such as ultrasonography and CT.9 This emphasises the utility of PET scanning in IgG4-RD. However, histology remains crucial for the diagnosis of IgG4-RD. The histological abnormalities should meet the Boston consensus on IgG4-RD.10 The characteristic histological features of IgG4-RD are dense lymphoplasmacytic infiltrate, storiform fibrosis and obliterative phlebitis. The ratio of IgG4/IgG-positive plasma cells in tissues should be greater than 0.4 and the numbers of IgG4-positive plasma cells per high power field should be greater than the numbers agreed in the consensus.10 The absolute numbers of IgG4-positive plasma cells and the thresholds for disease differ for the diverse organs. Our patients had histologically confirmed IgG4-RD according to the criteria; however, in case B no obliterative phlebitis was seen.
The pathogenesis of IgG4-RD is unclear.11 Generally, the disease is characterised by a decreased T-helper cells 1/T-helper cells 2 ratio and increased numbers of regulatory T-cells, most probably as a result from a certain antigen triggering the immune system. Production of different cytokines such as interleukin (IL)-4, IL-5, IL-10, IL-13 and transforming growth factor (TGF)-beta leads to co-activations of B-cells, production of IgG4 expressing B-cells and fibrosis. Still, the role of IgG4 antibodies is unclear, but in the pathophysiology of IgG4-RD these antibodies most probably play an anti-inflammatory role as response to an unknown trigger.12 Patient C presented with constrictive pericarditis and pleural effusion. Plasma cell manifestation of the pericardium has also been described in multiple myeloma,13 whereby infiltration of plasma cells in the pericardium is suggested to be the reason. Maybe some viral infection led to IgG4-positive plasma cell infiltration in the serosal cavity leading to the clinical manifestation of this disease, but this remains a speculative hypothesis. The pleural effusion was most probably also because of infiltration by lymphoplasmacytic cells, as it was slightly positive on PET and disappeared after starting prednisone. However, secondary pleural effusion because of restricted heart function due to constrictive pericarditis could also have contributed to the development of pleural effusion.
IgG4-RD can cause significant morbidity and even lead to organ damage. Aggressive treatment is therefore necessary, especially when vital organs are at risk.11
Glucocorticoids are the first choice of treatment for most types of IgG4-RD and are mostly effective at a prednisone dosage of 30-40 mg/day and should be adjusted according to body weight or in cases of aggressive disease.14 This treatment dose is, in most cases, rapidly effective, but should be maintained for two to four weeks after initiation. Thereafter, prednisone can be tapered according to clinical responses. The clinical response of prednisone is dependent upon the organ system involved and the degree of fibrosis. Pancreatic function and lacrimal gland function, for example, will respond better to this treatment than retroperitoneal disease or sclerosing mesenteritis.14 This phenomenon highlights the need for earlier treatment of this disease.5 About 25% of patients demonstrate relapse after tapering prednisone necessitating steroid-sparing agents. Patient A responded very well to prednisone. His symptoms, serum IgG4 and MRI imaging normalised and remained so during tapering. Patient C also responded very well to prednisone. His symptoms disappeared, serum IgG4 reached almost normal levels and a recent chest X-ray no longer demonstrated pleural effusion. Therefore, we decided not to initiate maintenance therapy in cases A and C. According to international consensus, a steroid-sparing agent is appropriate when the glucocorticoid dosage cannot be tapered due to persistently active disease.14 For this reason, azathioprine was initiated in case B. Conventional steroid-sparing agents such as mycophenolate mofetil, azathioprine and methotrexate have all been used in the treatment of IgG4-RD, but management of further immunosuppressive therapy with these disease-modifying antirheumatic drugs (DMARDs) has not been outlined14 and there are no studies confirming the superiority of one of these agents in the treatment of IgG4-RD. There is improving evidence for the efficacy of rituximab in the treatment of IgG4-RD, even as a single therapy.15 This B-cell ablative therapeutic agent has induced clinical remission in patients with various organ involvement of IgG4-RD.3 More case series or prospective studies with different DMARDs and rituximab are required in order to define the (long-term) effect of these agents in the treatment of IgG4-RD.
CONCLUSION
In conclusion, IgG4-RD is a rare and new clinical entity with many faces and manifestations in different parts of the body. Early recognition is critical to start treatment and to avoid permanent damage of the organs. Diagnosis is based on histology, while serum IgG4 could be supportive. Glucocorticoids are the first choice of treatment, but there is often a need for maintenance therapy. Several DMARDs as well as rituximab are used in the treatment of IgG4-RD, with growing evidence for the latter.
DISCLOSURES
This research was partly funded by Combined Ophthalmic Research Rotterdam grant 3.3.0.
The authors declare no conflicts of interest regarding the publication of this paper.
REFERENCES