KEYWORDS
Metabolic disorder, inborn error of metabolism, hyperammonaemia
INTRODUCTION
The urea cycle involves a series of biochemical steps in which nitrogen, a waste product of protein metabolism, is converted into urea. This urea is subsequently removed from the body by excretion in the urine. Urea cycle defect is a genetic disorder that results in a deficiency of one of the six enzymes of the urea cycle. These disorders all result in accumulation of nitrogen in the form of ammonia, leading to hyperammonaemia. High serum levels of ammonia are especially toxic for the brain, resulting in irreversible brain damage, coma and death. In late-onset ornithine transcarbamylase deficiency (OTCD) the genetic defect leads to partial enzyme deficiency. Under normal circumstances residual activity makes patients feel generally well. They may remain unrecognised. But in a catabolic state, ammonia levels can rise quickly causing imminent coma and death.
CASE REPORT
A 67-year-old male was admitted to our hospital because of progressive dyspnoea. He had a history of non-specific interstitial pneumonia (NSIP), for which he had never needed treatment with corticosteroids. On arrival, respiratory failure was established and the patient was transported to the ICU for intubation and inotropic support.
An exacerbation of NSIP was considered most likely, possibly combined with a superimposed infection. He was treated with high doses of prednisone, ceftriaxone and erythromycin.
After a few days he seemed to improve due to this treatment and the prednisone was decreased. However, a week after admission his pulmonary condition worsened dramatically. After thorough clinical, radiological and microbiological assessment, a focus of infection could not be found. Since a relapse of NSIP after decreasing corticosteroids seemed most obvious, methylprednisolone 1000 mg was given for three days.
After a short period of relative stabilisation the patient unexpectedly developed generalised seizures with signs of autonomic dysregulation. A cranial CT showed no pathology, neither was there any indication of meningitis or electrolyte disturbances.
After consultation of the neurologist, phenytoin and valproate were started, and when there was no improvement levetiracetam was added. Despite these measures the seizures increased in frequency and intensity. His pupils became dilated and unresponsive to light. Another CT scan, 12 hours after the first, showed a drastic change: diffuse cerebral oedema and signs of herniation, still without any indication of an intracerebral cause. Electroencephalography confirmed the clinical diagnosis of brain death.
In parallel, laboratory analysis was performed in search of a metabolic cause of the seizures. This revealed a strikingly high ammonia level of 2700 µmol/l (7-42 µmol/l). When repeated, analysis confirmed the result. The extensive differential diagnosis of hyperammonaemia1 was taken into account. After hepatic failure was excluded (normal liver enzymes, albumin, prothrombin time/international normalised ratio and echography) we considered an inborn error of metabolism.
Urine analysis demonstrated a strikingly high excretion of uracil 84.8 mmol/mol creatinine (0-15.1 mmol/mol creatinine), orotic acid 11.6 nmmol/mol creatinine (0-1.2 mmol/mol creatinine) and uridine 9.1 mmol/mol creatinine (0-3.7 mmol/mol creatinine). This is suggestive of a urea cycle defect. Genetic testing was performed in his two daughters, since there was no suitable blood sample available from the patient. We found that both were carriers of an OTC gene mutation (c.622G>A, p.Ala208Thr), a mutation more often described in OTCD patients.2,4 Later, screening revealed one of them to have passed the mutation on to her infant son. Also, family history revealed this mutation was found earlier in two distant cousins, genetically linked through the patient’s grandmother (figure 1). These findings prove that the cause of the patient’s fatal hyperammonaemia was late-onset OTCD.
In retrospect, other clues of OTCD were present. The patient had a tendency to avoid eating meat. He experienced light-headedness after consumption. This voluntary protein avoidance is characteristic for patients with a urea cycle defect.2 Also, his brother had ‘the flu’ at the age of 15, developed a coma and died a few days later.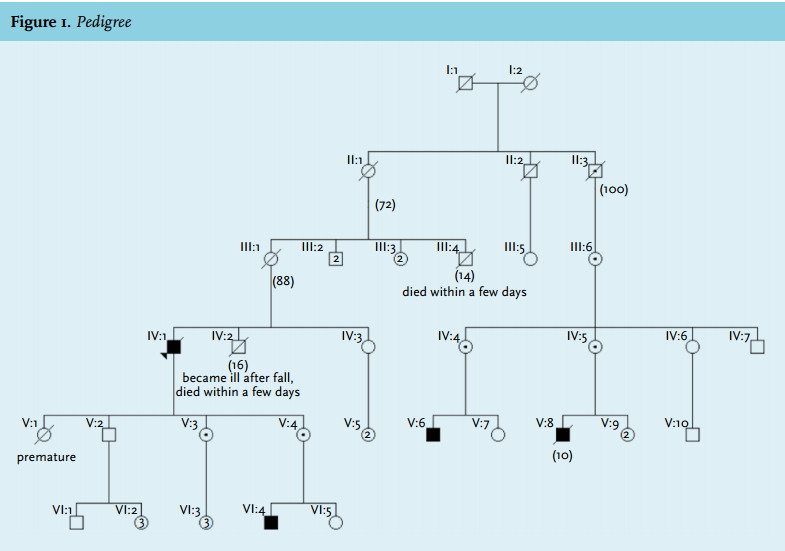
DISCUSSION
Ammonia is mainly produced in the gut, by enterocytes from glutamine or by bacterial degradation of urea and other nitrogenous substances, such as ingested protein. Another source is protein catabolism and muscle exercise.1 In the liver, ammonia is converted to carbamyl phosphate, which enters the urea cycle to be converted to urea. Urea is incorporated into amino acids or excreted in the urine. One of the enzymes required for the urea cycle is ornithine transcarbamylase (figure 2).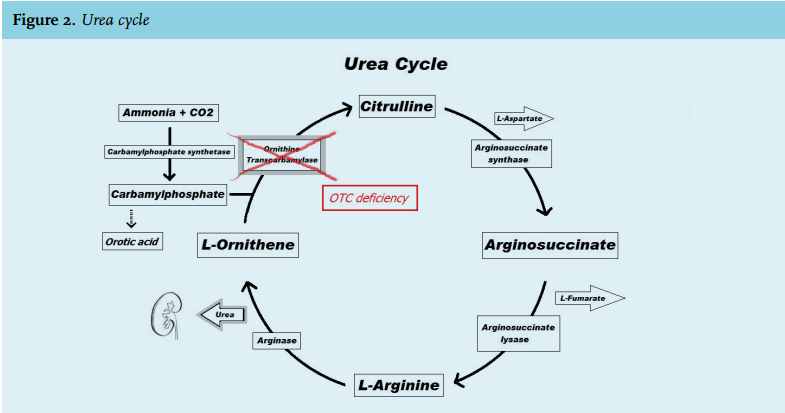
When the liver is unable to metabolise ammonia, elimination is carried out by the muscles, kidneys and brain. In the brain ammonia is metabolised to glutamine. Excessive amounts of intracerebral glutamine cause increased osmotic pressure and cerebral oedema.1
One of the causes of hyperammonaemia is a urea cycle defect. Ornithine transcarbamylase deficiency (OTCD) is the most common urea cycle defect in adults.2 It is an X-linked disorder due to deleterious mutations in the OTC gene. In hemizygous males the phenotype depends on the type of mutation - 50% have no residual enzyme activity at all and develop hyperammonaemic coma and death in the neonatal period. When some residual activity is present symptoms develop when environmental factors that luxate hyperammonaemia are present (late-onset OTCD). Female carriers are often asymptomatic since the phenotype is influenced by the X-inactivation pattern.
There are many environmental factors that may luxate hyperammonaemia in late-onset OTCD.1,3 With regard to our patient: he was in a catabolic state due to his illness, infection and prolonged ICU stay. He was given high-dose glucocorticoids, which are known to have a general catabolic effect primarily by enhancing protein turnover. There are at least four case reports of patients with OTCD who developed hyperammonaemic coma following steroid administration.2 Furthermore, he received protein-enriched enteral feeding in accordance to ICU guidelines, resulting in an increased nitrogen burden. Lastly valproate can cause hyperammonaemia, in patients with defect in the urea cycle, even at therapeutic dosages through inhibition of carbamyl phosphate synthase.1 However, the patient’s seizures had already developed before administration of valproate so the ammonia levels were already neurotoxic.
The relevance of recognising this disorder lies in the reversibility of hyperammonaemia and prevention of permanent neurological damage and death.4 Genetic counselling of all relatives is mandatory.
When a urea cycle defect is suspected, determination of plasma amino acids (ornithine, glutamine, alanine, citrulline), acylcarnitines (from blood or plasma), urinary organic acids and orotic acid is recommended.3 After recovery from an acute episode, plasma amino acid levels and/or urinary orotic acid can be helpful. Molecular genetic analysis is generally performed on DNA from the blood. RNA analysis from a liver biopsy is only carried out when DNA analysis is negative.3
Treatment should not be delayed, since the prognosis is strongly influenced by the duration of coma and peak ammonia levels.3,5
In the acute phase, protein intake should be stopped and a high caloric intake guaranteed to prevent catabolism (use dextrose 10% and intralipids). Ammonia should be removed, in severe cases by haemodialysis,6 otherwise by using ammonia scavengers (e.g. sodium benzoate, sodium phenylacetate). Supplementation of L-arginine, L-citrulline and L-carnitine improves ammonia excretion. The only curative treatment is liver transplantation; however, this has only rarely been performed in clinical practice and does not reverse sustained brain damage.
Chronic treatment consists of specialised dietary advice on protein restriction and optimal caloric intake. Catabolism should be prevented by all possible means. In addition ammonia scavengers, suppletion of citrulline and/or arginine can prevent hyperammonaemia.
In conclusion, we advise to obtain ammonia levels in all patients with unexplained coma, seizures or cerebral oedema. Lastly, voluntary protein avoidance could be one of the clues to the diagnosis of OTCD.
DISCLOSURES
No conflicts of interest for all authors. No funding. No prior abstract publication/presentation.
REFERENCES