
KEYWORDS
Acquired angioedema, allogeneic stem cell transplantation, treatment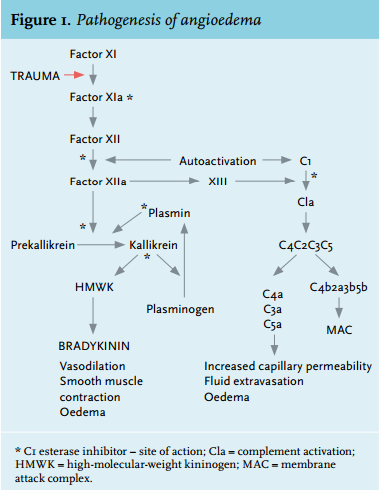
INTRODUCTION
Acquired angioedema is a rare disorder, characterised by recurrent attacks of non-itching, self-limiting subcutaneous oedema (or angioedema) and can present with life-threatening airway obstruction or with abdominal symptoms that mimic an acute abdomen, as upper respiratory tract and gastrointestinal tract are sites that are most often affected. Deficiencies in the inhibitor of the first component of human complement (C1-Inh) leading to angioedema can be either acquired or hereditary.1
In acquired angioedema the activity of C1-Inh is decreased and, subsequently, serum complement factors 4 (C4) and 1q (C1q) are low. Due to decreased levels of C1-Inh there is a continuous autoactivation of C1 leading to unrestrained activation of the classical pathway of the complement system. C1-Inh also inhibits factor XIIa and kallikrein, proteases belonging to the contact pathway. Increased vascular permeability due to massive bradykinin release via the contact pathway is thought to be the primary cause of symptoms in acquired angioedema (figure 1).2
Mechanisms causing acquired C1-Inh deficiency have been broadly investigated. Historically, acquired angioedema was defined as a constellation of syndromes, due to the many associated conditions such as lymphoproliferative diseases, systemic lupus erythematosus, primary myelofibrosis, autoimmune haemolytic anaemia, cryoglobulinaemia and liver hydatidosis.3 In 1986, Jackson et al.4 first described an IgG autoantibody inactivating C1-Inh demonstrating an autoimmune mechanism to be the cause of the C1-Inh deficiency in otherwise healthy patients. Acquired angioedema was divided into two types: type I, associated with lymphoproliferative disorders developing anti-idiotyping antibodies directed against the M-component. This immune complex causes activation of C1 and consumption of C1-Inh. Type II is primarily due to direct autoantibodies, impairing the activity of C1-Inh. Now it is becoming clear that there is an overlap between these two groups, since there are patients with lymphoproliferative diseases who also have anti-C1 inhibitor antibodies.5- 7 Thus, the division of acquired angioedema into these two types has been abandoned. Subsequent work revealed that acquired angioedema usually arises in the setting of an uncontrolled clonal proliferation of B lymphocytes. However, the mechanism by which clonal B cell disorders lead to depletion of C1-Inh and angioedema remains incompletely understood.
CASE REPORT
A 57-year-old male presented to our hospital with recurrent swelling of the hands, lips, tongue, scrotum and throat. He had no allergies, no urticarial rash, and was not on any medication known to provoke angioedema. Physical examination was normal. Laboratory tests showed the presence of an IgM kappa monoclonal antibody (2.76 g/l), with decreased activity of C1-Inh and consistent C1q and C4 consumption. CT scan of the thorax and abdomen showed no abnormalities. Bone marrow examination showed an increase in B-lymphocytes (24%), which were monoclonal. Immunophenotyping showed a monoclonal B lymphocyte population of 6%, positive for CD19, CD20, CD22, CD79a and expressing IgM kappa, compatible with a lymphoplasmacytoid malignant lymphoma (also frequently called Waldenström’s disease, however this term is not part of the current lymphoma classification). Additional analysis showed that in the IgM fraction autoantibody activity against C1-Inh was present (Professor M. Cicardi, Milan). This confirmed the diagnosis of acquired angioedema as a result of an IgM antibody directed against C1-Inh (type II).
Treatment was started with danazol and tranexamic acid. During the subsequent years his symptoms worsened, requiring high-dose purified human C1-Inh (15,000 units Cetor®/month). Despite this treatment he developed recurrent episodes of life-threatening laryngeal swelling, necessitating artificial ventilation. The M-protein was stable and there were no signs of osteolytic lesions or lymphadenopathy. Several courses of immunosuppression (high-dose prednisone, cyclophosphamide, azathioprine, mycophenolate mofetil and rituximab) were given with temporary relief of symptoms but recurred during maintenance therapy with prednisone, danazol and low-dose immunosuppression with prophylactic infusion of high-dose purified C1-Inh. In fact, he showed an increase in episodes of life-threatening laryngeal oedema. Two courses of rituximab, cyclophosphamide, vincristine and prednisone were given, again without a decrease in symptoms. C1-Inh activity at that time was only 7%. To treat the IgM-producing clone directly, we performed a non-myeloablative alloSCT, with his HLA-identical brother as donor. Awaiting the transplantation, he received two courses of rituximab, fludarabine, cyclophosphamide and mitoxantrone, again without response. Pre-transplant conditioning consisted of fludarabine and total body irradiation. The post-transplantation course was without complications. Immunosuppression consisted of mycophenolate mofetil until day +85 and tacrolimus until day +180. At day +100 chimerism was 97% donor and he was free of symptoms. To date (now year +5 after transplant) he is still in clinical remission with a C1-Inh activity of 75% and no clinical symptoms. There are no signs of graft versus host disease.
DISCUSSION
Here, we show successful treatment of acquired angioedema with a non-myeloablative alloSCT. The patient had very frequent and severe attacks of angioedema requiring high doses of Cetor®. After transplantation there was complete resolution of symptoms. Hereby, we provide evidence that alloSCT can suppress the antibodyproducing clone resulting in an increase of C1-Inh activity with a subsequent resolution of symptoms. Whether this patient is cured of the underlying M-protein producing clone remains to be seen since C1-Inh activity has not yet completely normalised.
Acquired angioedema is frequently associated with lymphoproliferative disorders, developing anti-idiotyping antibodies and/or autoantibodies. Autoimmunity and lymphoproliferation are closely connected in acquired angioedema. Different forms of B cell disorders coexist and/or evolve into each other, and seem to be dominated by alterations in the control of B cell proliferation. Blocking autoreactive or neoplastic B cell proliferation is probably essential in the treatment of acquired angioedema in these cases.2 Levi et al. 8 and Ziakas et al. 9 both described patients who were cured after treatment with anti-CD20 monoclonal antibody rituximab. There are data available supporting the efficacy of alloSCT in IgM-producing lymphoplasmacytic lymphoma. Kyriakou et al.10 described the outcome of alloSCT in 86 patients using either myeloablative (MAC; n = 37) or reduced-intensity conditioning (RIC; n = 49): the three-year non-relapse mortality rate was 33% for MAC and 23% for RIC, five-year overall survival rate was 62% for MAC and 64% for RIC and five-year progression-free rate was 56% for MAC and 49% for RIC. Gilleece et al. 11 described the outcome of alloSCT in the setting of lymphoplasmacytic lymphoma patients in the UK. In nine allografted patients transplantrelated mortality at 12 months was 44%.
Acquired angioedema can be treated with androgenic steroids, such as danazol. The underlying mechanisms are not yet clear, but it is believed to work either by stimulating the production of C1-Inh in the liver, increasing levels of aminopeptidase P, an enzyme that inactivates kinins, or by additional undetermined mechanisms.12 Antifibrinolytics, such as tranexamic acid, inhibit the conversion of plasminogen to plasmin and also inhibit activated plasmin. These steps in fibrinolysis are also inhibited by C1-Inh (figure 1).13 Administration of purified C1-Inh is also used in the treatment of acute attacks of acquired angioedema. Patients with acquired angioedema usually need much higher doses than patients with hereditary angioedema (20 units/kg).7 The bradykinin B2-receptor antagonist icatibant is suggested to be of value, based on its characteristic to antagonise the bradykinin B2 receptor (figure 1).14,15 At the time of treatment of this patient, no published data were available on the use of icatibant in patients with acquired angioedema. In addition, as we consider this patient to have a type 1 disease, we choose to treat the underlying disease by performing a non-myeloablative alloSCT.
In conclusion, to our knowledge, this is the first case to be described of acquired angioedema that was successfully treated with alloSCT. Five years after transplantation the patient is still in complete remission with no episodes of angioedema. Although further studies are warranted, this indicates that alloSCT may be a treatment option for acquired angioedema in case other known treatment options fail.
DISCLOSURE
The authors have declared that they have no conflict of interest.
REFERENCES