KEYWORDS
Boceprevir, direct-acting antiviral agents, hepatitis C, HIV, pegylated interferon-alfa, telaprevir
INTRODUCTION
The introduction of direct-acting antiviral agents (DAA) for the treatment of hepatitis C virus (HCV) infection heralded a whole new era of treatment possibilities with agents directed at multiple HCV targets (i.e. NS3, NS5A and NS5B).1 However, the speed of these developments has not been balanced by reimbursement policies which differ among countries.2 While in some countries interferon-free regimens with sofosbuvir and daclatasvir or simeprevir are now the standard of care, in many others first-generation protease inhibitors telaprevir and boceprevir are still the only available DAAs. Moreover, most middle- to high-income countries restrict the use of these new DAAs to those most in need: patients with advanced fibrosis, cirrhosis or extrahepatic manifestations.
Addition of boceprevir or telaprevir to peginterferonalfa (pegIFN-alfa) plus ribavirin has led to increased sustained virological response rates (SVR) of 65-75% for HCV mono-infected patients.3,4 Since HCV and the human immunodeficiency virus (HIV) are partly transmitted along similar routes, coinfection frequently occurs ranging from around 30% in men having sex with men to as high as 80% in injection drug users.5 In the Netherlands, around 10% of HIV-infected patients are coinfected with HCV.6 In these HIV/HCV coinfected patients, SVR rates with pegIFN-alfa and ribavirin have traditionally been lower compared with those achieved in HCV mono-infected patients.7,8 To date, only two small phase-2 studies and three cohort studies have been published showing similar efficacy of boceprevir and telaprevir in combination with pegIFN-alfa/ribavirin in HIV/HCV coinfected patients when compared with HCV mono-infected patients.9-13 However, varying rates of adverse events and treatment discontinuations due to differences in study populations (i.e. numbers of patients with cirrhosis and Caucasians) were reported in HIV/HCV coinfected patients.14 Furthermore, these cohort studies suffered from selection bias since mostly patients from early access or compassionate use programs were included. Extending the knowledge on treatment outcomes of boceprevir- and telaprevir-based therapy in HIV/HCV coinfected patients remains important for two reasons. First, despite the introduction of more novel direct-acting agents, boceprevir and telaprevir are still used in many countries around the world due to financial restrictions. Second, to confirm that boceprevir and telaprevir have similar effectiveness in HIV/HCV coinfected patients compared with HCV mono-infected patients. Here, we report the Dutch experience with boceprevir and telaprevir in a cohort of HIV/ HCV coinfected patients.
METHODS
Patients
From the Netherlands ATHENA HIV observational cohort registry, maintained by the HIV Monitoring Foundation (HMF),15 we selected all HIV-positive patients coinfected with chronic HCV genotype 1 of 18 years and older who were treated for 12 weeks or more with pegIFN-alfa/ ribavirin plus telaprevir or plus boceprevir between August 2010 and April 2013. Clinical visits included in this study were start of treatment (week 0), week 4, week 8 (for those with a pegIFN-alfa/ribavirin lead-in before boceprevir), week 12, week 24, week 48 (end of treatment) and week 12 follow-up (to assess SVR12, i.e. HCV RNA undetectable 12 weeks after the end of treatment). Data on sociodemographic characteristics and on HIV/HCV-related and haematological parameters together with reasons for treatment discontinuation were obtained from the registry. All patients within the ATHENA cohort gave consent (via an opt-out procedure) for their anonymised data to be collected and stored in a central database as part of their routine HIV care. A research proposal to perform the current study was submitted to and approved by the HMF working group.
Treatment
The standard duration of treatment was 48 weeks, while shortening to 24 (28 with boceprevir) or 36 weeks occurred based on clinical criteria at the treating physician’s discretion. Triple therapy was given with pegIFN-alfa 2a (180 µg weekly) or 2b (1.5 µg/kg weekly) together with weight-based ribavirin 1000-1200 mg daily (in two divided doses). Boceprevir was dosed orally at 800 mg three times a day (TID) taken with food for a duration of 24 to 44 weeks after a 4-week pegIFN-alfa/ ribavirin lead-in phase.16 Telaprevir was administered orally at doses of 750 mg TID in most patients while one patient received 1125 mg twice daily (BID) and another patient took 1125 mg TID because of a combination with efavirenz.17 Futility rules and treatment duration were as prescribed by the package insert of boceprevir and telaprevir, and in accordance with international treatment guidelines.18-20 Severe liver fibrosis was defined as F3 or F4 by METAVIR classification on preceding liver biopsy or by liver stiffness measurement (Fibroscan, Echosens, Paris, France), using a cut-off value of 12.5 kPa or higher.
HCV RNA determination and definitions of response
Plasma HCV RNA was quantified at the local hospitals with their respective polymerase chain reaction assay (COBAS Ampliprep/COBAS TaqMan V2.0, Roche Nederland B.V., Woerden, the Netherlands: detection limit 15 IU/ml; Abbott RealTime HCV, Chicago, USA: detection limit 12 IU/ml). A rapid virological response (RVR) was defined for telaprevir as an undetectable HCV RNA at week 4 of triple therapy whereas RVR for boceprevir-based therapy was defined by HCV RNA undetectability at week 8 of treatment.
Non-response to telaprevir was defined as plasma HCV RNA > 1000 IU/ml at week 4 or 12 during treatment while non-response to boceprevir was defined as plasma HCV RNA > 100 IU/ml at week 12 or detectable HCV RNA at week 24 of treatment. Relapse for both treatments was concluded when HCV RNA became detectable after being undetectable at the previous measurement.
Response to previous (pegylated) interferon-alfa/ ribavirin therapy was defined by classical definitions for non-response/ relapse as published in international guidelines.18,20
Statistical analysis
Data were analysed using descriptive statistics with continuous variables expressed as median with interquartile range, and categorical variables as numbers with percentages. Mann-Whitney test was used for continuous variables while Fisher’s exact and Kruskal-Wallis test was performed for categorical variables. An intent-to-treat analysis was used calculating loss to follow-up, deceased or discontinuation due to adverse events as treatment failures. The primary endpoint of this study was SVR12. Data were analysed using Graphpad Prism V5 for Mac (San Diego, California, USA).
RESULTS
Study population
A total of 53 HIV/HCV coinfected patients, 45 men and 8 women, were included in this study (table 1) with a median age at the start of HCV treatment of 47 years (IQR 44-56). Two-thirds of the cohort were infected with HCV genotype 1a and 26% had severe liver disease (F3 or F4). Regulative duration of treatment differed for telaprevir and boceprevir, which was also apparent in our study cohort. Telaprevir was prescribed for a median duration of 12 weeks (IQR 12-13 weeks) with pegIFN-alfa/ribavirin continuing until a median of 48 weeks while boceprevir was prescribed for 43 weeks (IQR 24-44) after a 4-week lead-in with pegIFN-alfa/ ribavirin. Raltegravir was more frequently used in patients on boceprevir compared with those on telaprevir (86% versus 54%; p = 0.03). Finally, there was no difference in HCV genotypes and baseline HCV RNA between boceprevir- and telaprevir-treated patients.
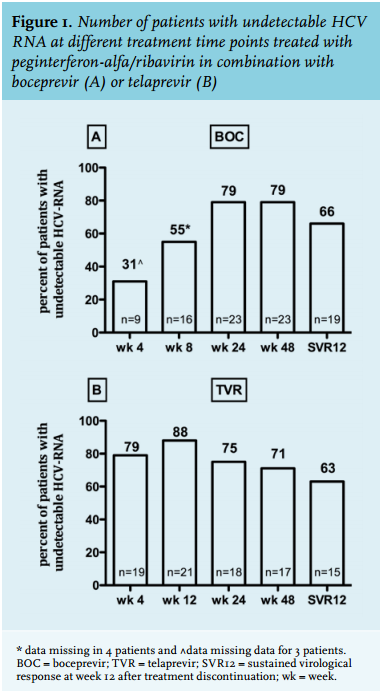 Treatment response
Treatment response
SVR12 in boceprevir- and telaprevir-treated patients is depicted in figure 1A and 1B and was achieved by 19 of 29 (66%) and 15 of 24 (63%) patients, respectively. The difference at week 4 of therapy between patients on telaprevir and on boceprevir (HCV RNA week 4 undetectable in 79% and 31% respectively) is explained by the pegIFN-alfa/ribavirin lead-in phase in the latter group (figure 1A and B). Virological non-response, breakthrough and relapse rates are shown in table 2. Four patients treated with boceprevir and three on telaprevir had a primary non-response on treatment. In the telaprevir group, one patient was lost to follow-up and another patient died 16 weeks after the start of treatment. Although both patients achieved an RVR, according to the intention-totreat principle, they were regarded as non-responders and treatment failures. Relapse rates were 17% (n = 5) in the boceprevir- and 9% (n = 2) in the telaprevir-treated group (p = 0.44).
Predictors of treatment response
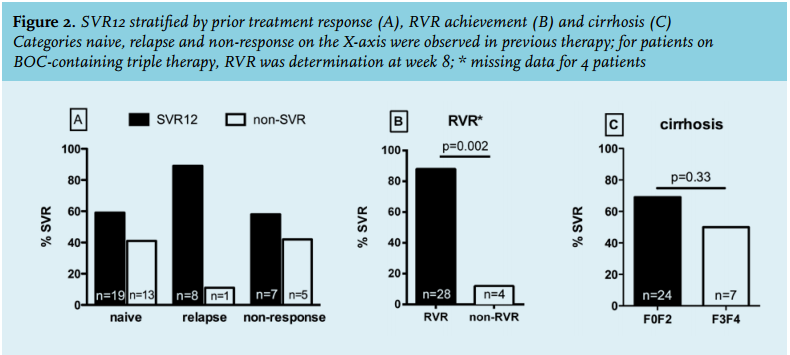
Since patient characteristics and treatment outcomes of boceprevir- and telaprevir-treated patients were comparable, we analysed predictive factors for treatment success in the overall study population. Patients with relapse after a previous course of pegIFN-alfa/ribavirin therapy had the highest SVR12 rates of 89% (8 out of 9 patients) followed by previous non-responders (64%; 7 of 11) and HCV-therapy naïve patients (59%; 19 of 32) (figure 2A). RVR was achieved in 35 of 53 (66%) patients of whom 28 (88%) went on to achieve an SVR12 (p = 0.002) (figure 2B). In contrast, four patients without an RVR still managed to reach SVR12. Sixteen of 25 (64%) boceprevir-treated patients and 19 of 24 (79%) telaprevir-treated patients achieved an RVR (p = 0.35).
Treatment was shortened in eight of 35 patients with an RVR, two on boceprevir (treated for 28 and 36 weeks) and six on telaprevir (median 24 week (IQR 21-27). Of those, six (75%) subsequently went on to achieve an SVR12 while two patients (one on boceprevir treated for 28 weeks and one on telaprevir treated for 24 weeks) experienced a relapse. Presence of severe liver fibrosis or cirrhosis did not markedly influence treatment outcome with seven of 14 (50%) of F3-F4 patients reaching SVR12 compared with 24 of 35 (69%) with F0-F2 reaching SVR12 (p = 0.33) (figure 2C). Finally, other factors such as HCV genotype, baseline HCV RNA, CD4 cell count and ethnic origin were not associated with treatment outcome (data not shown).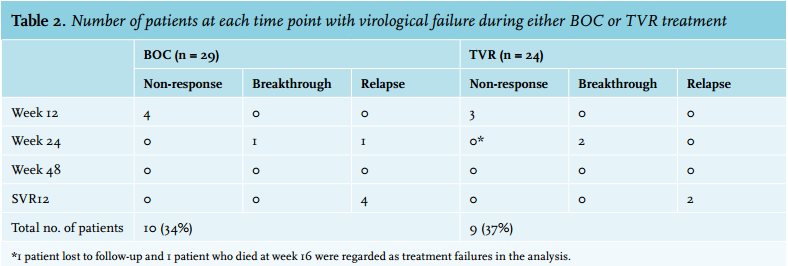
Safety of treatment
Boceprevir- and telaprevir-based therapies were generally well tolerated with recorded symptoms being malaise, diarrhoea and dizziness. Except for two patients, no severe adverse events leading to treatment discontinuation were noted in the ATHENA database. One patient was hospitalised for reasons not related to the telaprevircontaining triple therapy and subsequently achieved an SVR12. Another patient with Child-Pugh C (MELD-Na score of 40), baseline platelets of 75 x 109/l and an albumin level of 28 g/dl, died 16 weeks after the start of treatment with pegIFN-alfa/ribavirin and telaprevir due to complications of liver cirrhosis (i.e. spontaneous bacterial peritonitis with subsequent hepatic encephalopathy and hepatorenal syndrome).
Haemoglobin measurements at baseline, week 4 and week 12 were available in 48 patients (89%). The median baseline haemoglobin was 9.2 mmol/l (14.8 g/dl), which dropped to a median of 1.4 mmol/l (0.6 g/dl) after 4 weeks and 2.7 mmol/l (4.3 g/dl) after 12 weeks, respectively (p < 0.0001). Erythropoietin was prescribed in four (8%) patients while no patients discontinued therapy because of severe anaemia. Dose reduction of 200 mg ribavirin daily was done in six patients including those on erythropoietin therapy.
Two patients (one on boceprevir and one on telaprevir) experienced severe anaemia which in one patient contributed to non-compliance, resulting in viral breakthrough, while the other patient decided to stop therapy with subsequent viral relapse (table 2). Two other patients, both treated with telaprevir and achieving an SVR12, experienced leukopenia for which a pegINF-alfa dose reduction was applied.
Despite adequate antiviral drug concentrations, one patient developed HIV viral breakthrough at week 16 during anti-HCV therapy. His antiviral regimen was changed from atazanavir/ritonavir/raltegravir/maraviroc to darunavir/ritonavir/maraviroc with subsequent HIV RNA undetectability. However, at week 24 of anti-HCV therapy his HCV RNA was again detectable (i.e. relapse). All other patients were HIV undetectable during the course of their anti-HCV therapy.
DISCUSSION
The outcome of boceprevir- and telaprevir-based triple therapies in HIV/HCV coinfected patients in ‘real-life’ is favourable and these results are comparable with SVR data previously obtained in clinical trials and early access programs in both HCV mono-infected and HIV/HCV coinfected patients.3,4,10,11 Furthermore, this study again confirms that patients with a relapse on previous (peg) IFN-alfa/ribavirin therapy have a high chance of achieving treatment success on these triple therapies.21,22 This latter observation has consistently been reported among other boceprevir- or telaprevir-treatment studies without a clear explanation. It could be that since previous relapsers on pegIFN-alfa/ribavirin therapy have been exposed to these modalities, they know what to expect from treatment and therefore have a better tolerance or adherence.
The phase-3 clinical trials with boceprevir or telaprevir in combination with peg-IFN-alfa/riba performed in HIV/ HCV coinfected and HCV mono-infected patients reported comparable SVR12 rates of around 65%-75%.3,4,10,11 This is higher than previously achieved with pegIFN-alfa/ribavirin therapy with SVR24 rates between 17-36% for HCV genotype 1 in HIV/HCV coinfected patients.23-25 In contrast, with the high efficacy of new interferon-free regimens with around 90% SVR rates there is no difference in outcome (SVR12) between HIV/ HCV coinfected and HCV mono-infected patients.26 However, efficacy of triple therapy reported from early access and real-life cohorts in HIV/ HCV coinfected patients varied due to differences in included patients. For example, the CUPIC cohort and other more recently published cohorts reported SVR12 rates between 40%-55% for boceprevir and telaprevir in treatment-experienced and/ or cirrhotic HCV mono-infected patients.27-29 Other studies have, however, reported higher SVR rates of around 61%-80% in similarly affected HIV/HCV coinfected patients.9,13 Our study is distinctive since it describes a relatively healthy, in majority HCV therapy naive, population with only a small proportion of cirrhotic patients. Moreover, the Dutch HIV healthcare system is concentrated in a few specialised treatment centres with highly trained infectious diseases and HIV nurses. This might explain the low number of severe side effects and low drop-out rate seen in our cohort. However, one patient with cirrhosis died after reaching 16 weeks of therapy while being HCV RNA undetectable. This patient’s baseline platelet count and albumin were 75 x 109/l and 28 g/dl, respectively, which in the CUPIC cohort were found to be associated with an increased risk of death.27 On this basis, triple therapy with either boceprevir or telaprevir is contraindicated in those patients with a low albumin and low platelet count. Furthermore, probably due to a relatively small sample size, treatment outcome was not statistically significantly affected by fibrosis stage though a difference in percentage was notable (50% in F3-F4 versus 69% in F0-F2). This is in line with the literature showing that the presence of liver cirrhosis is a negative predictor for outcome of DAA-based therapy.30
Considering the long duration of triple therapy in combination with many described side effects of therapy, shortening of therapy might be a possibility in some patients with favourable HCV viral kinetics. Although the number of patients in whom shortening of therapy was performed (at the treating physician’s discretion) was small, a favourable outcome especially for those on telaprevir was seen in this study. Similarly, shortening triple therapy from 48 weeks to 24/28 weeks was recently also investigated in the HIVCOBOC-RGT study by Mandorfer et al. 31 Although the number of patients in the study was small, a 100% SVR12 rate was reached in those 14 becoming HCV RNA undetectable (‘target not detected’) at week 8 of therapy (i.e. including the four-week lead-in phase). Moreover, in our study there was one patient on telaprevir lost to follow-up after 16 weeks of therapy who was regarded as a treatment failure. However, this patient had a favourable viral kinetic response with HCV RNA undetectability at week 2 of telaprevir-based triple therapy. Several publications have shown that very short courses of triple therapy are sufficient to achieve an SVR.32,33 In all, shortening of therapy based on RVR undetectability with similar SVR rates and lower costs of therapy might be a favourable strategy, especially in resource-limited setting.
There are some limitations to this study. Since we collected our data from the Dutch HIV database, certain data regarding severity of fibrosis such as albumin and platelets were not collected the way data were collected in the CUPIC cohort. Finally, there are small differences (though not statistically significant) in baseline characteristics such as fibrosis stage and prior treatment response between patient groups treated with either telaprevir or boceprevir. However, when analysing the data excluding the four patients without data on fibrosis stage, the SVR12 rate dropped to 60%, only marginally lower than for the whole study population. We therefore think that these differences in baseline characteristics did not influence the outcome in this study.
In conclusion, SVR12 rates were favourable for pegIFN-alfa/ribavirin with boceprevir or telaprevir in this relatively healthy cohort of HIV/ HCV coinfected patients and comparable with those in HCV mono-infected patients. Furthermore, although numbers were low, shortening of treatment duration seems feasible in those patients who achieve HCV RNA undetectability at week 4 of therapy.
DISCLOSURES
J.E. Arends: Advisory boards of Janssen, MSD, Abbvie, ViiV, BMS; Speakers Bureau Gilead.
K. Brinkman: Advisory boards of Janssen, MSD, Abbvie, ViiV, BMS, and Gilead.
C. Smit: The Athena cohort is supported by an institutional subsidy from the Dutch Ministry of Health, Welfare and Sport and was set up and is maintained by the Stichting HIV Monitoring.
M. van der Valk: Consultancy Abbvie, BMS, Gilead, Janssen, Roche and research support from MSD and Janssen.
P. Reiss: through his institution has received independent scientific grant support from Gilead Sciences, Janssen Pharmaceuticals Inc., Merck&Co, Bristol-Myers Squibb, Boehringer Ingelheim and ViiV Healthcare. In addition he serves on a scientific advisory board for Gilead Sciences and on a data safety monitoring committee for Janssen Pharmaceutica N.V., for which his institution has received remuneration
C. Richter: advisory board of MSD, BMS, Janssen, Roche and AbbVie.
A.I.M. Hoepelman: Consulting honorarium Janssen, Advisory boards of Janssen, Gilead, BMS, MSD and ViiV.
The other authors declare no conflicts of interest.
REFERENCES