KEYWORDS
Myelopathy, systemic lupus erythematosus, neuropsychiatric lupus erythematosus

INTRODUCTION
Neuropsychiatric manifestations, ranging from headache or mild cognitive confusion to seizure disorder or myelopathy, affect up to 80% of patients with systemic lupus erythematosus (SLE) and are associated with considerable morbidity and mortality.1 Myelopathy, one of the neuropsychiatric SLE syndromes (NPSLE) that have been described by the American College of Rheumatology (ACR) (table 1), is a rare but clinically important condition, due to the dramatic presentation with rapidly evolving paraparesis, sensory deficits and sphincter dysfunction.2 The clinical decision-making process in these patients can be challenging as a result of the broad differential diagnosis, and the urgency to initiate treatment early in the course of the disease to improve prognosis.3,4 This case report is aimed at the recognition of myelopathy in SLE patients and outlines the most important clinical challenges in these patients.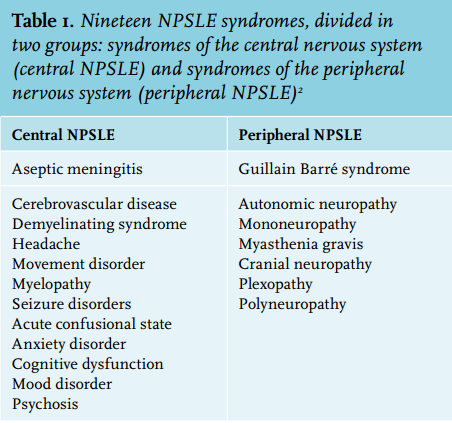
CASE REPORT
A 29-year-old female of Creole descent with a history of non-inflammatory arthralgias in the ankles and wrists (2009) and a transient psychosis during the third trimester of her first pregnancy, for which she was briefly treated with haloperidol (2011), developed paraparesis and urine incontinence two months after her first childbirth. Medical history was otherwise unremarkable. On physical examination, grade II paresis of the lower extremities (according to the Medical Research Council scale) was found with normal sensory functions, diminished quadriceps and gastrocnemius reflexes and absent Babinski’s sign. Laboratory results showed an elevated erythrocyte sedimentation rate of 119 mm/h (reference range 1-20 mm/h), leukopenia of 3.5 x 109/l (4.0-10.0 x 109/l) and normocytic anaemia of 5.1 mmol/l (7.5-10.0 mmol/l). Apart from a positive IgG Coombs test, there were no signs of active haemolysis, based on a normal bilirubin of 8 μmol/l (reference range < 17 μmol/l), normal reticulocytes of 38 x 109/l (25-120 x 109/l) and a high haptoglobin of 2.8 g/l (0.3-2.0 g/l). Other laboratory results, including electrolytes and vitamin B12, were normal.
Myelopathy was suspected, and a broad differential diagnosis was formulated, including compressive causes (e.g. trauma or neoplasms) and non-compressive causes (e.g. demyelination, infections, inflammatory or vascular disorders, toxic or metabolic abnormalities and paraneoplastic syndromes).5
Additionally, CT and MRI imaging of the brain and spine were performed, which were both normal. Cerebrospinal fluid (CSF) examination was not suggestive of infections of the nervous system, with a normal opening pressure, mild leukocytosis (9.0 x 106/l; reference range < 5.0 x 106/l), elevated protein levels (2.8 g/l; 0.2-0.5 g/l), low glucose (2.3 mmol/l; 2.8-4.0 mmol/l) and elevated IgG (789 mg/l; 0.0-34.0 mg/l) with an increased IgG index (0.68; 0.30-0.60) and oligoclonal IgG band. CSF cultures were negative.
Serological testing for human immunodeficiency virus and cytomegalovirus was negative. Electromyography and motor evoked potential were both normal, thereby ruling out peripheral nerve disease, plexopathy and neuromuscular junction disease as an explanation for the patient’s paraparesis and urine incontinence.
At this point, we excluded compressive causes, demyelination, and infectious diseases as an explanation for the patient’s myelopathy. The diagnostic process focused on inflammatory disorders, supported by the patient’s history of arthralgias and concomitant leukopenia, anaemia and elevated ESR. The differential diagnosis of inflammatory myelopathy was further subdivided into SLE, sarcoidosis and antiphospolipid antibodies syndrome. A more detailed history revealed no other autoimmune phenomena.
We performed additional laboratory testing, which revealed the presence of antinuclear antibody (ANA), anti-doublestranded DNA (anti-dsDNA) with a titre of 15 kU/l, anti-ribonucleoprotein and anti-smooth muscle antibodies. Complement levels were elevated. A recent study has demonstrated that complement levels and anti-dsDNA are unreliable markers for NPSLE: only one-third of patients with NPSLE had low complement levels and positive anti-dsDNA during a symptomatic phase.6 Diagnostic work-up for antiphospholipid syndrome was negative (absent lupus anticoagulant, anti-cardiolipin antibodies and anti-β2 glycoprotein I antibodies).
Sarcoidosis was deemed unlikely due to an unremarkable chest X-ray (no hilar lymphadenopathy or interstitial abnormalities) and the absence of skin or eye abnormalities. Subsequently, the diagnosis SLE (psychosis, leukopenia, positive ANA and anti-ds DNA) with SLE-associated myelopathy was made.
Intravenous treatment with methylprednisone (1000 mg for three days) and cyclophosphamide (1300 mg, 750 mg/m2 ) was started when she developed bilateral Babinski’s signs, after which her symptoms quickly improved.
DISCUSSION
Myelopathy, affecting 1-2% of SLE patients, can be divided into two subtypes.7 Firstly, transverse myelopathy, which is more prevalent in SLE, with involvement across one level of the spinal cord. Secondly longitudinal myelopathy in which more than four levels of the spinal cord are affected, either continuously or separately.8 The most common presentation of patients with transverse myelopathy consists of paresthesias (usually at level T5-T8), which can be accompanied by paraparesis and/ or sphincter dysfunction.4 A small subgroup of patients may have normal sensory functions.4
In patients suspected of SLE-associated myelopathy, MRI is the preferred neuro-imaging technique and may detect T2-weighted hyperintense lesions, the most common finding in NPSLE.7,9 In 54-93% of these patients abnormal lesions are found.4 The meaning of these lesions is uncertain, although they may be indicative of microinfarcts, focal ischaemia, demyelination, or gliosis.10 Importantly, they correlate poorly with clinical manifestations and are aspecific for NPSLE. Similar lesions can be found in non-SLE patients and in SLE patients without neuropsychiatric manifestations.10
CSF examination and cultures should be performed in order to exclude infectious diseases. In 50-80% of NPSLE patients mild, non-specific abnormalities (e.g. pleocytosis, elevated protein levels and decreased glucose levels) are found.7 An elevated IgG index and oligoclonal bands, indicative of an immunological response, may be found in patients with SLE but are also found in other diseases, such as multiple sclerosis and sarcoidosis.13
As soon as infectious causes are excluded or SLE-associated myelopathy is suspected, treatment with immunosuppressive drugs should be initiated. Preferably, initial treatment should consist of intravenous methylprednisone for three consecutive days followed by intravenous cyclophosphamide (750 mg/m2 ). This regimen should be followed by monthly infusions of cyclophosphamide for 6-12 months, combined with oral prednisolone 1 mg/ kg for three months, which should be tapered afterwards according to disease activity.4,14 In NPSLE patients with refractory symptoms, treatment with rituximab (anti-CD20 monoclonal antibody) can be considered.15 Next to immunosuppressive therapy, anticoagulation has been suggested as a therapeutic option for SLE patients with aPL-positive myelopathy.7 However, a systemic review failed to show any additional effect of anticoagulation on the neurological outcome of 70 patients with aPL positive myelopathy.16 Anticoagulant therapy has not yet been evaluated in a randomised, placebo-controlled setting. 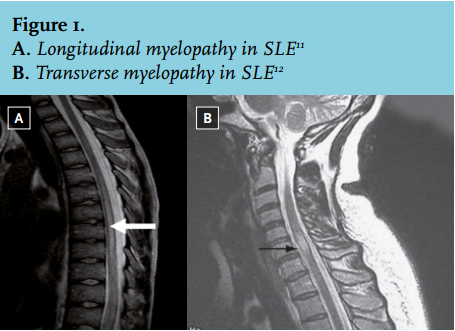
Our patient was aPL-negative and therefore not treated with anticoagulant therapy. She showed clinical improvement after the start of immunosuppressive therapy but still had a grade IV paresis in the lower extremities after discharge. For four consecutive months, during which she received monthly cyclophosphamide infusions, her disease remained in remission. Due to side effects, cyclophosphamide was changed to oral azathioprine, which successfully maintained the remission.
The overall outcome of patients with SLE-associated myelopathy seems reasonably favourable.4 Complete recovery occurs in up to 50% of the cases, whereas 21-31% of the patients show no clinical improvement or deterioration after immunosuppressive therapy.
In conclusion, the diagnostic work-up in patients suspected of SLE-associated myelopathy, consisting of MRI and CSF examination, is vital in order to rule out the presence of other underlying diseases. Thereafter, aggressive immunosuppressive therapy with methylprednisone and cyclophosphamide should be initiated early in the course of the disease to favourably affect outcome. Clinical trials should be initiated to evaluate the current treatment regime, including the role of anticoagulant therapy, for SLE-associated myelopathy.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES