KEYWORDS
Haemoglobinopathy, liver transplantation, bilirubin, Mizuho, Gilbert, hyperbilirubinaemia
INTRODUCTION
A patient with a known rare haemoglobinopathy was admitted with symptoms of fulminant hepatic failure requiring liver transplantation. He appeared to have a combination of two interacting genetic mutations. Molecular diagnostic possibilities have grown enormously in the past decades. As a consequence, more rare inherited diseases are diagnosed. Due to this, knowledge about these diseases is difficult to obtain, even with modern literature availability. Syndromes previously diagnosed based on clinical criteria can now be diagnosed even if the symptoms are obscured by concomitant diseases. We describe a case report of a patient with two rather benign single nucleotide polymorphisms (SNP); one very rare and one more common, but in combination leading to severe complications.
CASE PRESENTATION
A 25-year-old male was transferred from another hospital to our intensive care unit (ICU) with acute liver and renal failure to evaluate the possibility of acute liver transplantation. He had been diagnosed by DNA analyses with Mizuho haemoglobin as a child,1 which is a rare disease of which only three other patients have been described in the world.2-4 In this de novo heterozygous T→C mutation there is a Leu→Pro substitution at position 68 of the β-chain. This type of haemoglobin has higher oxygen affinity and the β-chain is unstable. It is associated with chronic haemolysis. A cholecystectomy was performed for symptomatic cholelithiasis at the age of 4 and a splenectomy at the age of 6 in an attempt to reduce haemolysis. After this last operation his transfusion need diminished. During his entire life the transfusion load was approximately 40 units of concentrated red blood cells with the majority given before the splenectomy. Until then his total and mostly conjugated bilirubin levels were around 200 µmol/l, due to which his jaundice was striking. He had no other signs of cholestasis such as itching. He led an active life and worked as a hiphop journalist. At the age of 21 he showed progressive jaundice. Endoscopic retrograde cholangiopancreatography (ERCP) revealed stenosis of the bile duct. A balloon dilatation of the common bile duct with stenting was performed, resulting in a decrease in the bilirubin levels to pre-existing values. The same year the stent was removed without any effect on the cholestasis. Also, he was diagnosed with pT3N1bM0 papillary thyroid carcinoma at the age of 23, for which he was curatively treated with a total thyroidectomy plus lymph node dissection and radioactive iodine therapy.
Two weeks before admission to our ICU, he presented himself in another hospital with complaints of fatigue and progressive jaundice. These symptoms had slowly progressed over the months before. ERCP and sonography showed no signs of bile duct obstruction. Revision of the liver biopsy performed in 2012 showed intrahepatic cholestasis with some portal and parenchyma fibrosis, without iron or copper overload. During his admission, he developed coagulopathy and visual disturbances and a few days later also renal failure. Because there was a suspicion of acute hepatic failure with hepato-renal syndrome requiring a liver transplant, he was transferred to our ICU.
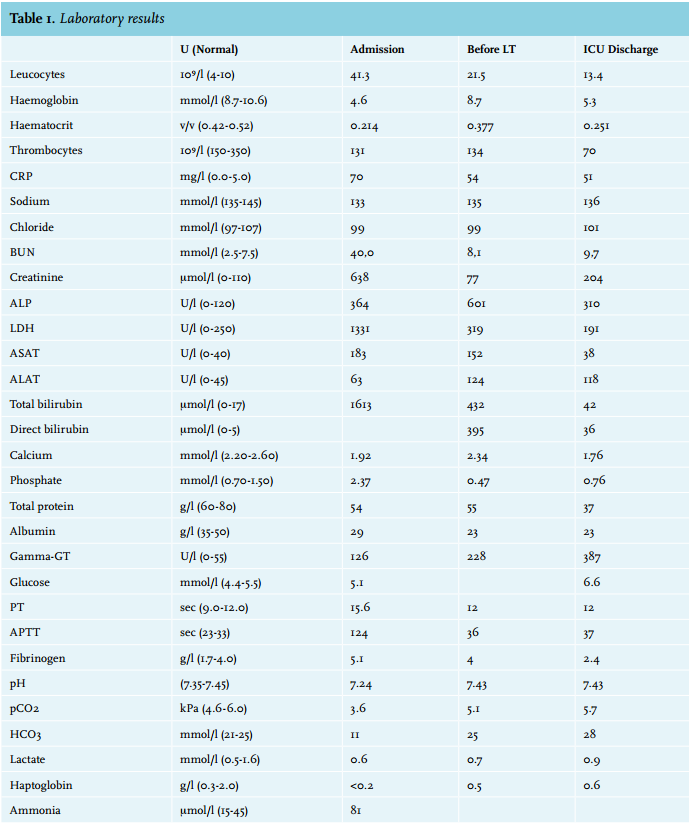
On admission, his total bilirubin level was 1613 µmol/l and almost entirely conjugated, haemoglobin was 4.6 mmol/l, his activated partial thromboplastin time (APTT) was 124 seconds and his creatinine level was 638 µmol/l (table 1). Encephalopathy grade 2 was present.5 Autoimmune and viral serology revealed no aetiology for his clinical deterioration. A sonography of liver and heart showed no major abnormalities. The diagnosis fulminant hepatic failure of unknown origin was made. On admission he already met the Kings College criteria for acute liver transplantation and he was placed on the transplantation waiting list.5,6 To bridge time to transplantation, molecular adsorbent recirculation system (MARS) therapy combined with continuous veno-venous haemofiltration (CVVH) was started. This quickly improved his liver and renal function and the patient also improved clinically. His neurological symptoms disappeared. The request for a donor liver was put on hold temporarily, because of his improved liver synthesis as measured by coagulation parameters. After four days he could be discharged to the ward, at which time his total bilirubin levels were 516 µmol/l and his coagulation tests were normal.
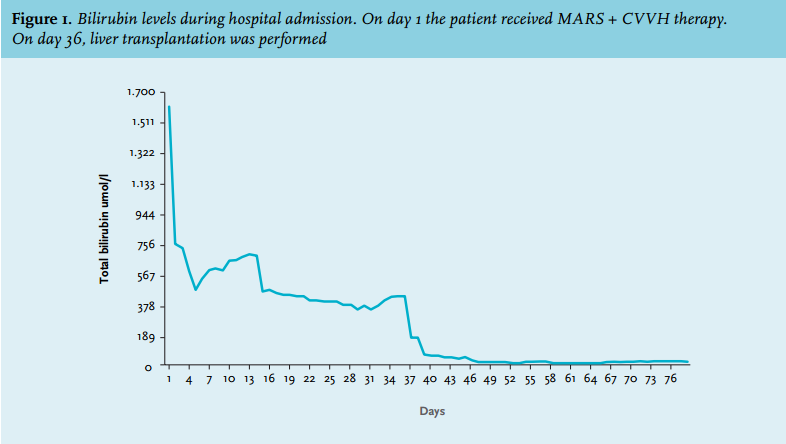
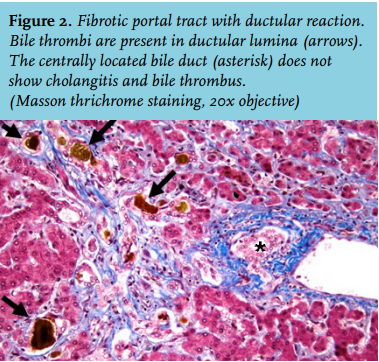
After six days the patient was readmitted to the ICU because he had a fever, pain on his left flank and hypotension. Also, his bilirubin levels were increasing again and he developed extreme pain in his right upper quadrant. A liver biopsy showed massive intrahepatic cholestasis with porto-septal fibrosis but also peri-sinusoidal and centrilobular fibrosis, without signs of a primary and/or obstructive biliary disease. Three days later he developed progressive hypotension and bradycardia. Echocardiography now showed a large pericardial effusion, causing a tamponade. A pericardiocentesis was performed and haemorrhagic fluid was drained. There was improvement for a few hours but then again he developed hypotension followed by cardiac arrest. An emergency thoracotomy was performed in the ICU, which resulted in a return of spontaneous circulation after removal of blood and clots from the pericardium. Three days later, he again developed a tamponade and a re-thoracotomy was performed in the operating room. No micro-organisms were cultured from the pericardial fluid. For the entire period in the ICU, his haemoglobin levels were kept > 9 mmol/l to suppress erythropoiesis and subsequent haemolysis. CVVH was continued for renal failure. Bilirubin levels remained around 400 µmol/l. Magnetic resonance cholangio-pancreatography to exclude bile duct obstruction again showed no abnormalities. Four days after the last thoracotomy the request for a donor liver on the high urgency list was reactivated. Eleven days later he underwent a liver transplantation and received a full-size liver from a heart beating donor. Two days after that a re-laparotomy was performed because of intra-abdominal haemorrhage. Shortly hereafter the patient stabilised: his total bilirubin level dropped directly postoperatively to 162 µmol/l and ten days later it was 26 µmol/l (figure 1). CVVH could be discontinued because of the return of spontaneous diuresis. Intermittent haemodialysis was started. One week after liver transplantation he was discharged to the ward. The histology of the explanted liver showed massive cholestasis with bile thrombi in dilated canaliculi, in ductular and ductal lumina and bile sludge in the larger ducts without features of cholangitis. There were numerous foci of groups of hepatocytes with feathery degeneration, a feature of hepatocyte loss due to severe cholestasis. The liver showed diffuse septal fibrosis with porto-portal and porto-central bridging and also sinusoidal fibrosis. Months later genotyping revealed that the patient had Gilbert’s syndrome as a second SNP. Meanwhile, the patient was doing well and he could perform his job again. His bilirubin levels stayed within normal limits.
DISCUSSION
This patient was transferred to our hospital with the clinical diagnosis of fulminant hepatic failure of unknown origin, but probably related to his rare haematological condition. This disease is called Mizuho after the Japanese residence were the first patient was described. Very little is known about this disease because there are only four cases reported in the literature, all without long-term follow-up; one describes our patient.1-4 After splenectomy this disease seems to have a relatively benign course.1-4 In this patient there was no severe haemolysis as measured by lactate dehydrogenase. MARS was started as a bridge to transplantation.7 After that, the bilirubin levels halved and the patient improved considerably. Encephalopathy and coagulation disorders disappeared and the synthesis function of the liver appeared normal. Liver transplantation was postponed to await further improvement. But then, rising bilirubin levels and pericardial tamponade urged further therapy. He was again placed on the transplantation list under the diagnosis of chronic haemolysis with unknown underlying hepatic disease leading to bilirubin toxicity. Liver damage due to chronic cholestasis, both hepatocellular and in intra- and extra-hepatic bile ducts, was considered an alternative. However, liver biopsy did not reveal a specific diagnosis. Genetic tests were ordered but the results were still pending at the moment of transplantation.
Our hypothesis was based on the literature. The main issue and ultimately the clue was that this patient with a relatively low-grade haemolysis showed this level of jaundice with most of the bilirubin conjugated. This conjunction of laboratory findings could not be fitted in a single known syndrome as fulminant hepatic failure, bile duct obstruction, haemolysis or any other. There are several inherited diseases with chronic or intermittent haemolysis with a higher incidence than Mizuho. These diseases have a widely differing geographical distribution but are far more common than Mizuho.
Sickle cell disease (SCD) and thalassaemia are also haemoglobinopathies. Furthermore, glucose-6-phosphate dehydrogenase (G-6-PD) and spherocytosis are metabolic disorders of the erythrocyte that lead to haemolysis. Sickle cell anaemia and thalassaemia can lead to liver failure.8,9 SCD is associated with hepatopathy.10 Liver failure in SCD is, next to complications of sickling, due to transfusion-related infections and iron overload.11-14 Sickle cell intrahepatic cholestasis (SCIC) is an extremely rare complication of SCD.15 The clinical presentation of SCIC includes severe abdominal pain, acute hepatomegaly, coagulopathy, extreme hyperbilirubinaemia and acute hepatic failure, which can also lead to renal failure. For adults this is a potentially fatal process.13,16-18 In children it sometimes resolves spontaneously.19 The cause of SCIC is unknown but in the literature the presumed pathogenesis is that sickling red blood cells plug the hepatic sinusoids causing cholestasis and local hypoxia.12,13,16 Hyperbilirubinaemia is mainly conjugated in contrast to hyperbilirubinaemia due to haemolysis which is mostly unconjugated.13 Treatment consists of supportive care and prompt exchange transfusion to minimise haemolysis.10,13,16 However, this may not always lead to improvement. There are a few case reports of patients with SCIC who received liver transplantation. One recent article by Gardner et al. describes a patient with suspected SCIC in a chronic form, developing hepatic failure ten years later.13 The patient responded very well to liver transplantation and two years later he was still in a good condition. Most patients improve after exchange transfusion which not only removes sickling cells, but also a substantial amount of albumin-bound bilirubin.15,16 Except for the fact that our patient did not suffer from SCD, his clinical presentation strongly resembles SCIC. Moreover, the case of Khurshid et al. is the first one to also develop a tamponade with haemorrhagic fluid in the pericardium and later on brady-arrhythmias.15 The pericardial bleeding was probably caused by coagulopathy combined with heparin administration through CVVH. Extreme hyperbilirubinaemia is associated with renal failure and coagulopathy.17 Our patient has the extremely high levels of bilirubin in common, which may be a leading factor in disease development. Unlike in our patient, SCD causes chronic haemolysis with bouts during crises leading to high bilirubin.
The most robust genetic association that modifies the clinical phenotype of haemoglobinopathies is Gilbert’s syndrome; however, several others have been described.20-22 Beta thalassemia intermedia or major and sickle cell anaemia can co-occur with Gilbert’s syndrome and lead to increased bilirubin.23 Gilbert’s syndrome was described for the first time in 1901.24 The incidence of Gilbert’s syndrome in the general population is approximately 8%.25-27 There is racial variability.26,28-30 Gilbert’s syndrome is caused by a genetic defect in bilirubin conjugation with glucuronide. Mutations in the promoter region of UGT1A1 are the most frequent polymorphism.31 UGT1A1 promoter with 7 TA repeats in the promoter region has less enzyme activity than the most frequent genotype with 6 repeats.28 An increasing number of repeats decreases the transcription efficiency 20-fold.29,31 Our patient had 7 repeats. The diagnosis of Gilbert’s syndrome is usually based on unconjugated hyperbilirubinaemia without haemolysis, normal liver tests and normal ultrasonography of the liver.25,32 This makes the diagnosis complicated in patients with chronic haemolysis as in haemoglobinopathies.33
UGT1A1 (TA)7 is associated with higher bilirubin, cholelithiasis and cholecystectomy in SCD, β-thalasaemia, hereditary spherocytosis and G-6-PD.30,34-40 Our patient had gallstones and a cholecystectomy at the age of 4, which could have triggered the suspicion of a concomitant disorder if the similarity with other haemoglobinopathies had been recognised. As a child our patient suffered from hyperbilirubinaemia which was mainly unconjugated, as could be expected in both haemolysis and Gilbert’s syndrome. In this episode his hyperbilirubinaemia was mainly conjugated as has been described in SCIC with extremely high bilirubin.13 The explanation for this shift is hypothetical but it might be chronic toxicity of extremely high bilirubin.
For acute SCIC prompt exchange transfusion is the therapy of choice. But in extreme hyperbilirubinaemia MARS might have an important additional beneficial effect as shown in this patient. However, MARS clears the plasma compartment only. Our patient was deep dark green reflecting the loading of tissues with bilirubin, and this might explain the early relapse in our patient. Plasmapheresis in extreme hyperbilirubinaemia is also probably effective by removing albumin-bound bilirubin.15,41,42 Using the same strategy, looking for analogies we did not find an explanation for the thyroid carcinoma. Thus far we consider it to be a coincidence.
CONCLUSION
Molecular diagnostics revealed a very rare disease for this patient in his childhood. The unfamiliarity with this disease initially leads to the attribution of all symptoms to Mizuho’s disease. However, molecular diagnostics also made it possible to diagnose Gilbert’s syndrome, which is normally diagnosed on clinical grounds if there is no haemolysis. As with other rare and complicated diseases not all symptoms and his complete history can be explained. But this history illustrates that the phenotype of a genetic syndrome heavily depends on the rest of the genotype. It is important to always look for other causes if symptoms cannot be explained with the disease a patient is already diagnosed with. It also illustrates that in rare diseases the clue can sometimes be found searching in the immense available literature for analogy. In this patient liver transplantation could fortunately repair one of his diseasing polymorphisms.
ACKNOWLEDGEMENTS
We thank Dr J de Witte, Jeroen Bosch Hospital, ’s-Hertogenbosch, for providing the data about transfusion history. We thank Mr. B. Stevens for checking the English language and grammar as a native English speaker.
DISCLOSURES
The authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES