KEYWORDS
Diagnostic workup, epidemiology, maintenance treatment, non-cystic fibrosis bronchiectasis treatment
INTRODUCTION
Bronchiectasis – characterised by irreversible, pathological dilatation of the small and medium-sized bronchi – is not a disease in its own right, but rather a final common pathway of a vicious cycle of inflammation, bacterial colonisation and infection. A variety of respiratory and systemic diseases may be complicated by pathological bronchial dilatation, and therefore various medical specialists will be dealing with the condition in one way or another. Although general availability of computed tomography (CT) scans has importantly contributed to higher case-finding rates, bronchiectasis is still considered an underdiagnosed condition. In this article, we address the different signs and symptoms which can be clues to the diagnosis in order to facilitate recognition of the disease among non-pulmonary physicians. We further discuss our preferred diagnostic approach and give an overview of evidence-based treatment options.
Cystic fibrosis, an inherited multi-system disorder, is usually discussed separately and here we focus on non-cystic fibrosis bronchiectasis – hereafter referred to as ‘bronchiectasis’. The gold standard for diagnosis has long been bronchography, until the introduction of high-resolution CT scanning, the current standard diagnostic test. Due to the abundant amount of purulent phlegm produced by affected individuals, bronchiectasis was considered offensive and also untreatable before the introduction of antimicrobial agents.1
Around World War I, bronchiectasis was common in the Western world and it carried a poor prognosis: over 40% of all patients died of respiratory causes before the age of 40.2-4 Improved socio-economic status, successful nationwide vaccination programs for whooping cough and measles, and – most importantly – the availability of antibiotics reduced both incidence and mortality, in developed countries at least. Indeed, bronchiectasis became an ‘orphan disease’, as a result of which the focus of clinicians and researchers diverted away from this condition, which was now considered rare with a relatively benign course. In spite of adequate antibiotic treatment, however, bronchiectasis still has the potential to cause substantial morbidity, including repeated lower respiratory infections complicated by haemoptysis, a disabling productive cough and shortness of breath, all of which importantly affect quality of life.2 Patients with bronchiectasis were found to spend more days in hospital and have higher annual medical care expenditure as compared with matched controls.3 Recent epidemiological studies show a high incidence of bronchiectasis among New Zealand’s and Australia’s indigenous population and inhabitants of remote areas in Alaska.4 In the developed world estimated prevalence ranges from 0.42 per 100,000 in 18-34 year olds to 272 per 10,000 in those over 75.5
Important developments in the last decade include the introduction of international guidelines, the proposal for a validated scoring system for disease severity and the first large randomised trials on antibiotic maintenance treatment for those with frequent exacerbations, all of which will be discussed in this article.6-9 We illustrate the evidence from the literature on the diagnosis and treatment of bronchiectasis using the experience gained in a large Dutch teaching hospital. Demographic, epidemiological and clinical data were collected from the entire, unselected, non-CF bronchiectasis cohort of the Alkmaar Medical Centre in 2010, for research purposes. Data were retrieved from chart review of all adult patients with recurrent lower respiratory tract infections and high-resolution CT-proven non-CF bronchiectasis who visited the outpatient clinic for respiratory diseases of the Medical Centre Alkmaar at the time.
PATHOPHYSIOLOGY
The mechanism of disease that eventually causes bronchiectasis is traditionally depicted as a vicious circle of excessive inflammation and bacterial colonisation. Diverse stimuli, which can be either endogenic (such as ciliary defects) or exogenic (e.g. foreign body aspiration), may result in structural damage to the airways. This in turn allows for persistent bacterial colonisation of the larger and medium-sized bronchi. The host inflammatory responses together with secreted bacterial toxins cause additional damage (hypersecretion, ciliary dysfunction and airway remodelling) which further weakens local resistance.10,11
The immune response in bronchiectasis is mainly neutrophil driven and increased levels of chemokines and pro-inflammatory cytokines are found in the airways of affected individuals.12,13 High levels of proteases – toxic neutrophil products excreted on neutrophil activation – are present at the site of inflammation, resulting in release of pro-inflammatory cytokines and exerting proteolytic activity, thus causing even more damage to cells constituting the structure of the airways.14 T-cell infiltration, impaired macrophage phagocytosis, altered epithelial cell function and, more recently, deficiency of mannose-binding lectin have all been proposed as additional mechanisms responsible for an enhanced inflammatory response.11,15-18 A cycle of oxidative stress is also present, in which (mainly neutrophil derived) reactive oxygen species cause damage to cells and the surrounding tissues and induce additional oxidative stress through activation of the inflammatory transcription factors nuclear factor-kappa B and activator protein-1.19
CAUSES
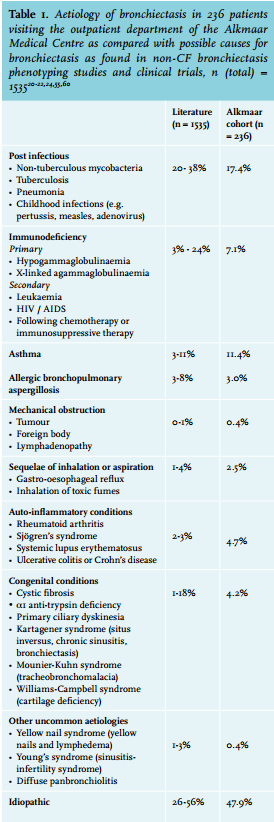
Bronchiectasis may arise from several different causes, headed by infection and immunodeficiency, mostly primary antibody deficiency syndromes (table 1). Due to successful prevention programs for tuberculosis and childhood infections such as whooping cough and measles, post-infectious bronchiectasis tends to become less common in developed countries. In about half of the patients, no underlying cause of permanent airway damage is found. Shoemark et al.20 found no causative factor in one third of their patients despite thorough systematic investigations in a tertiary referral centre. Other centres with multidisciplinary specialised bronchiectasis outpatient clinics with diagnostic protocols in place report 40-50% idiopathic bronchiectasis in spite of an extensive workup.21-24 Bronchiectasis is seen in 7-25% of patients with asthma or chronic obstructive pulmonary disease (COPD), coinciding with more severe disease.25,26 While asthma has recently been considered a cause of bronchiectasis in the absence of other factors, the link between COPD and bronchiectasis has yet to be established.6 The underlying cause for our cohort of patients is shown in table 1.
CLINICAL PRESENTATION AND SYMPTOMS
The ‘typical’ patient with bronchiectasis is supposedly a middle-aged woman, who is a lifelong non-smoker – or at least, this is the profile of the majority of patients in bronchiectasis phenotyping studies.20-23,27,28 Our own data do not completely reflect this picture, as our patients were slightly older and more frequently smokers (table 2). This incongruence illustrates the varied clinical presentation of bronchiectasis patients in clinical practice. Bronchiectasis can just as well occur in the 80-year-old male with frequent and virulent exacerbations of obstructive lung disease as in the 40-year-old lady with rheumatoid arthritis visiting your practice with complaints of persisting cough. Severity of symptoms is different for each patient, but in general the course of the disease is highly variable, including nearly symptom-free periods interspersed with infectious exacerbations. The most persistent and often presenting symptom is a chronic productive cough, present in 96% of 103 patients referred to a pulmonary outpatient clinic, with the amount of sputum being among the main determinants of quality of life.2 Dyspnoea, fatigue and upper respiratory tract symptoms are encountered in 60-70% of patients. About half of the patients describe having specks of blood in their sputum at any time, but haemoptysis resulting in immediate medical consultation is present in a quarter of patients. Pleuritic or musculoskeletal chest pain is present in 25-50% of patients and chest pain is often the reason for repetitive investigations at emergency departments. Exacerbations are characterised by an increase in symptoms and signs suggesting lower respiratory tract infection. Physical examination is often unremarkable except for the presence of crackles, mostly bilateral at the lower lobes.27,28
DIAGNOSTIC WORKUP
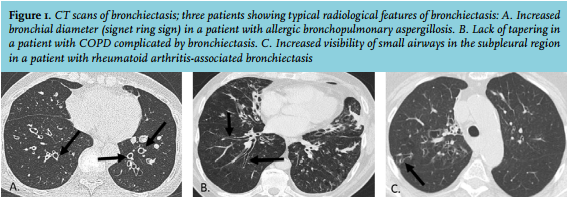
Bronchiectasis ought to be considered in patients with a chronic productive cough and/or recurrent lower airway infections, especially when these symptoms are present in younger, non-smoking individuals. Haemoptysis, recurrent para-nasal sinus infections or successive sputum cultures positive for S. aureus or P. aeruginosa may also be clues leading to the diagnosis. In patients with asthma or COPD, bronchiectasis should be considered in case of frequent, slow-resolving exacerbations, unstable or medicationresistant asthma or severe symptoms despite limited exposure to smoking in patients diagnosed with COPD.6 Key to the diagnosis are imaging studies using high-resolution CT. The chest CT protocol should be a spiral CT with 1 mm slices, able to detect pathology of larger and smaller airways, preferably with software allowing reconstruction in different planes. In patients with bronchiectasis, high-resolution CT typically shows a distorted ratio (> 1.0) of the inner bronchial diameter as compared with the accompanying artery, and signs of bronchial dilatation: lack of tapering and increased visibility of small airways in the sub-pleural region (figure 1).29 Plain chest X-rays show abnormalities in a large proportion of bronchiectasis patients (66% of our cohort), but changes are non-specific and an unremarkable chest X-ray does not rule out bronchiectasis.
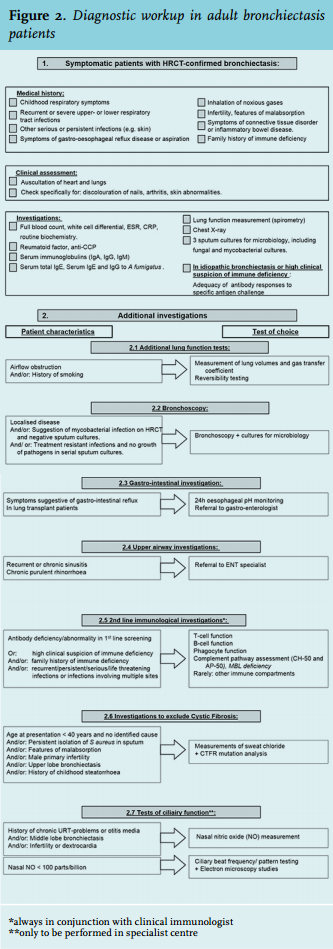
In symptomatic patients, the radiological finding of bronchiectasis should be followed by investigations to reveal the underlying cause. If a standardised protocol is used, the diagnostic yield may be enhanced, resulting not only in reduction of the proportion of patients diagnosed with ‘idiopathic’ bronchiectasis, but even in changing the treatment and the prognosis in up to 50% of patients.30,31 We use a diagnostic algorithm based on national and international guidelines (figure 2).6,32,33
A standardised workup has been shown to reduce diagnostic delay, which could last for up to several years, especially in patients with underlying immune deficiency.34 Localised bronchiectasis is usually indicative of a local mechanical cause (e.g. middle lobe syndrome) or post-infectious damage. The latter is even more plausible when a clear temporal relationship exists between an infectious episode and development of bronchiectasisrelated symptoms. In other subjects, bronchiectasis can occur as a symptom of an already identified disease, such as rheumatoid arthritis or inflammatory bowel disease. In such cases we suggest to refrain from extensive investigations – or to only resort to additional testing if unexplained deterioration occurs. The same holds true for patients with asymptomatic bronchiectasis, as for instance can be seen in stable fibrosis (traction bronchiectasis).
TREATMENT OPTIONS
When a specific disorder is found to cause bronchiectasis, disease management should primarily be directed at the underlying cause. This, for instance, applies to bronchiectasis due to allergic bronchopulmonary aspergillosis or common variable immune deficiency, both requiring their own treatment regimens. Bronchiectasis management is aimed at preventing disease progression and improving quality of life by reducing symptoms and exacerbations. This includes treatment of exacerbations and optimal airway clearance, complemented with long-term antibiotic therapy (oral or nebulised) or surgery in selected cases. Many treatment options for non-CF bronchiectasis are derived from the treatment regimens developed for cystic fibrosis. At first, treatment modalities were simply extrapolated to non-cystic fibrosis patients, but in the last decade, treatment modalities have been studied for this specific group of patients, resulting in evidence-guided treatment recommendations. Sometimes these recommendations contradict those for cystic fibrosis, as is true for mucolytic treatment with recombinant human DNAse (rhDNAse). Routinely used in cystic fibrosis treatment, rhDNAse was found of no benefit in one trial of non-CF bronchiectasis and harmful in another.35 Insufficient evidence is available to support the use of other mucolytics, such as acetylcysteine, in non-cystic fibrosis patients. Inhaled corticosteroids – although widely used by non-specialists in non-CF bronchiectasis patients – were only found effective in patients with underlying asthma. Current guidelines advise against routine use in non-CF bronchiectasis.6 It is worth mentioning that the pharmacological options described below – such as macrolides or inhaled hyperosmolar agents – have been approved by neither the US Food and Drug Authorization nor the European Drug Regulators. Use is solely based on outcomes of clinical trials and international guidelines.
Management of infectious exacerbations
One of the cornerstones of bronchiectasis management is antibiotic treatment of infectious exacerbations. There are no randomised trials evaluating the effect or the duration of antibiotic treatment in bronchiectasis, but antibiotics are generally thought to reduce the time to recovery and to reduce symptoms. By convention, a 14-day course of antimicrobials is prescribed, either intravenously or orally for exacerbations that last several days at least and are accompanied by increased sputum purulence, volume or reduced viscosity and increased cough, dyspnoea and systemic upset such as fatigue or fever.6 Preceding antibiotic treatment, sputum samples should be submitted for microbiological investigation and therapy should be directed at previously or newly isolated pathogens.
Physiotherapy
Most patients with bronchiectasis, especially those with excessive secretions, are offered physiotherapy. A customary physiotherapy program in the Netherlands would include one or more techniques directed at improved clearance of broncho-pulmonary secretions, combined with a pulmonary rehabilitation program to improve exercise tolerance. Forced expiratory manoeuvres as well as hand-held devices generating positive expiratory pressure (‘pep’ devices) such as Flutter™ or Acapella™ are used for optimal sputum clearance. A recent randomised trial, evaluating a similar approach, demonstrated a beneficial effect on exercise capacity, dyspnoea and fatigue in 85 patients.36 A Cochrane review, evaluating the effect of physiotherapytaught airway clearance techniques (ACT) as compared with no therapy or active coughing, demonstrated small improvements in sputum expectoration, lung function and health-related quality of life in five small and diverse studies, involving 51 patients.37 The choice of an ACT might as well be guided by patient preference, since there is no clear evidence in favour of any of the ACTs available. A small randomised study in 30 patients showed improved exercise tolerance and health-related quality of life with pulmonary rehabilitation in addition to ACT as compared with ACT alone.38
Inhalation of hyperosmolar agents
Due to impaired mucociliary clearance, many patients with bronchiectasis suffer from mucus hypersecretion and retention, leading to dyspnoea, chronic cough and increased susceptibility to infections. We frequently use inhalation of isotonic (0.9%) or hypertonic saline (6-7%) twice daily in addition to airway clearance techniques for optimal sputum evacuation. An evident benefit of nebulised hypertonic saline over isotonic saline has not yet been demonstrated in the small studies available and in our experience, patients report less discomfort in terms of wheezing or dyspnoea when using the isotonic solution.39 Nevertheless, the inhalation process itself is often experienced as time consuming and inconvenient. The hyperosmolar agent mannitol reduces exacerbations and improves lung function in cystic fibrosis.40 When administered as dry powder through a purposedesigned inhaler device, it is proposed as a less cumbersome alternative to saline inhalation. Several smaller or short-term studies on mannitol inhalations in bronchiectasis yield conflicting results in terms of sputum expectoration and quality of life.39 The sole large – yet slightly underpowered – long-term trial of 400 mg mannitol twice daily vs. a non-therapeutic dose of 50 mg demonstrated that inhaled mannitol increases the time until first exacerbation in patients with bronchiectasis, without improving respiratory quality of life or reducing actual exacerbation rates.41 Mannitol is known for inducing bronchospasm. It is worth noting that all participants in two large clinical trials were screened for mannitol tolerance at baseline and excluded when mannitolinduced bronchospasm was present (in 16% of all screened subjects). In the other participants mannitol inhalations were safe and well-tolerated.41,42 In the Netherlands, dry powder mannitol (Bronchitol™) is primarily used for optimising sputum expectoration in cystic fibrosis patients and is not registered for use in other patient groups.
Long-term antibiotic treatment
Treatment with maintenance antibiotics in bronchiectasis can be directed at simply reducing the increased bacterial load, since chronic colonisation has been found to coincide with enhanced inflammation and worse clinical outcome. In case of macrolides it is thought to dampen the exaggerated inflammatory response through multiple pathways.43
Macrolides
Macrolides, because of their anti-bacterial and anti-inflammatory properties, have long been thought ideal to intervene in the vicious circle of infection and inflammation that underlies bronchiectasis. In three different clinical trials evaluating long-term oral macrolide treatment, exacerbation frequency was significantly reduced. All trials used different dosing regimens and there is an ongoing debate on which schedule should be used. Traditionally, many physicians use a dosing schedule equivalent to the cystic fibrosis treatment schedules consisting of azithromycin 500 mg thrice weekly or 250 mg daily. Similar schedules were used in the BAT and EMBRACE trials, as opposed to the Australian BLESS trial which used erythromycin 400 mg twice daily.7-9 In cystic fibrosis patients macrolide antibiotics, and in particular azithromycin, tend to cumulate inside alveolar macrophages and as such have an extended half-life. Based on the pharmacokinetic properties of azithromycin in cystic fibrosis patients – whose kinetics may differ considerably from those without cystic fibrosis – dose levels of 22-30 mg/kg/week divided by 1-7 dosing moments, are proposed.44 Lung function improvement and enhanced quality of life were most distinct in patients with frequent exacerbations. Recent COPD trials show a tendency to a higher yield of macrolide treatment in patients with more exacerbations.45 Although bronchiectasis guidelines consider patients with three or more exacerbations yearly and suffering from chronic symptoms to be candidates for this treatment type, no robust evidence is as yet available to justify abstaining from macrolide treatment in less frequent exacerbators.7-9 Benefits of macrolide treatment come with a considerable increase in macrolide-resistant pathogens, which demands judicious use of long-term macrolide therapy.
Inhaled antibiotics
Since the late 1990s, nebulised antibiotics for reducing airway bacterial load have been considered a treatment option in bronchiectasis. Higher bacterial load is found to coincide with augmented systemic inflammation and increased morbidity.46 Due to the favourable pharmacokinetic profile of inhaled substances, with minimal systemic drug delivery, systemic adverse effects are mild.47,48 Local, non-severe side effects are frequently encountered in clinical trials with inhaled antibiotics.49 Inhalation-induced bronchospasm could pose an extra challenge in clinical practice, but is usually overcome through inhalation of a short-acting beta-2 agonist prior to inhalation of antibiotics. Most randomised clinical trials evaluating inhaled antimicrobial agents included bronchiectasis patients colonised with Pseudomonas aeruginosa and used different types of antibiotics (colomycin, tobramycin, amikacin, or ciprofloxacin).50-54 In addition, the three distinct trials (using aztreonam, gentimicin and ciprofloxacin), which did not specifically require P. aeruginosa colonisation for inclusion, in fact included many patients with P. aeruginosa colonisation at baseline (48-85%).49,55,56 All trials demonstrated bacterial load reduction in the airways of actively treated patients, but this effect does not correspond consistently with improvement in clinical endpoints.57 The largest trial (n = 500) of inhaled aztreonam in bronchiectasis patients – 85% of whom were P. aeruginosa colonised – failed to demonstrate reduced exacerbation rates or improved quality of life.49 Other authors report prolonged time to exacerbation and improved health-related quality of life as secondary findings. The attractive safety profile and encouraging results in some studies have stimulated further research in this field and momentarily no less than seven trials are recruiting patients, most of which studying inhaled ciprofloxacin.58 Awaiting further evidence we think that nebulised antibiotics offer a reasonable alternative to oral treatment in selected patients colonised with P. aeruginosa. Other non-pharmacological options, such as surgery for localised disease and bronchial artery embolisation in case of massive haemoptysis, will not be discussed here in detail.
PROGNOSIS
Although bronchiectasis can cause considerable morbidity, prognosis in terms of survival is favourable. The largest prospective study up until now found 62 deaths in 608 patients (10.2%) within four years, but the majority of deaths (81%) occurred above the age of 70.59 Independent predictors of mortality were older age, low FEV-1, prior hospitalisation and three or more exacerbations in the year prior to the study. The authors used these data to compile and validate a clinical prediction tool, the Bronchiectasis Severity Index, which divides patients into three risk groups (low/ intermediate/ high) in order to predict mortality, hospital admissions and exacerbations. This tool could be very useful in research settings in order to increase homogeneity of study populations. Its value for directing therapy in a clinical setting still needs to be proven.
In conclusion, the broad range of diseases that cause or coincide with bronchiectasis make it a frequently encountered entity in various medical specialisations. The authors hope that this article will renew awareness of this still underdiagnosed condition. Exciting new developments are the publication of high-quality, randomised studies and new tools for patient selection which are important steps towards improving bronchiectasis management.
DISCLOSURES
The authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES