KEYWORDS
Adults Still’s disease, Griscelli syndrome type 2, haemophagocytic lymphohistiocytosis, macrophage activation syndrome, systemic lupus erythematosus
BACKGROUND
Macrophage activation syndrome (MAS), a secondary form of haemophagocytic lymphohistiocytosis (HLH), is an underrecognised and life-threatening condition associated with a heterogeneous group of diseases including connective tissue diseases (CTD), cancer, infections, and exposure to drugs.1 Both MAS and HLH are characterised by inappropriate survival of histiocytes, cytotoxic CD8+ T cells, and macrophages, leading to a massive cytokine release, hyperinflammatory state and haemophagocytosis.2,3 Clinically, patients with MAS-HLH present with persistent fever, hepatosplenomegaly, cytopenia, hyperferritinaemia, coagulation disorders, elevated inflammatory markers (C-reactive protein (CRP) and soluble interleukin-2 receptor (sIL2R)), hypertriglyceridaemia, and histopathological evidence of haemophagocytosis and multi-organ failure.4 Recognition of MAS-HLH may be challenging since clinical features can mimic other conditions such as sepsis, shock, and underlying active systemic disease.5 MAS-HLH occurs more frequently in children than in adults, however, reporting in adults is increasing.1
Despite several studies reported on MAS-HLH and its association with different triggers and underlying diseases, the mortality remains high with rates of up to 80%, possibly also due to diagnostic delay.1,6,7 In this report, we describe three adult cases with different presentations of MAS-HLH and their clinical features and outcomes, aiming at improving early identification and treatment of MAS-HLH.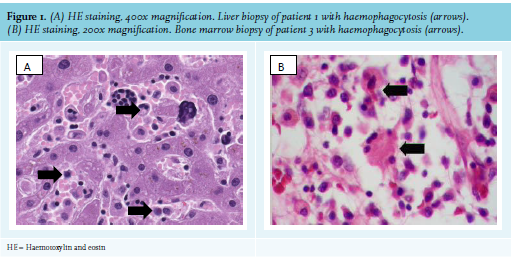
Case 1
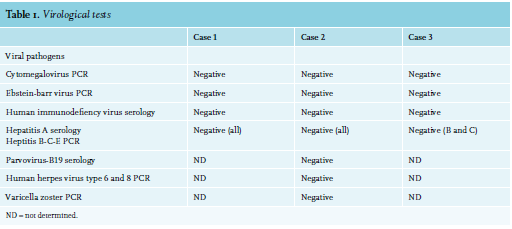
A 40-year-old female with a previous history of mild subacute cutaneous lupus erythematosus (SCLE) for the past 16 years, presented with fever, malaise, photosensitivity, and SCLE flare. Treatment with prednisone 60 mg daily initially resulted in improvement of cutaneous lesions. However, the patient developed jaundice and laboratory tests revealed deteriorated cholestatic liver tests with elevated transaminases, as well as haemolytic anemia (11 g/dl), thrombocytopenia (86 cells/ul), leucopenia (3600 cells/ul), and hyperferritinaemia (807 ng/ml). Physical examination revealed a fever of up to 40°C and diffuse macular erythematous rash. Autoantibody, viral screens, blood and urine cultures were negative (table 1).
Abdominal ultrasound examination showed hepatosplenomegaly without biliairy obstruction. A skin biopsy revelaed erythema multiform, however, SCLE flare could not be ruled out. The liver biopsy was initially interpreted as drug-induced liver injury.
During follow-up, her clinical condition worsened with respiratory insufficiency, progressive haemogram, and liver test disturbances (see table 2 for maximum values). The patient was admitted to the intensive care unit (ICU). Progressive loss of renal function with concurrent haematuria and proteinuria developed. Complement 3 and 4 levels were decreased. Revision of the liver biopsy showed signs of haemophagocytosis (figure 1). Bone marrow biopsy revealed neither malignant cells nor haemophagocytosis. Renal biopsy showed acute tubulus injury and bile cast nephropathy.
A diagnosis of MAS-HLH secondary (19 days after initial presentation) to SLE was established and treatment with ciclosporin and pulse methylprednisolone was initiated. Ciclosporin was discontinued due to nephrotoxic effects and treatment with anakinra was started. After initiation of anakinra, liver tests improved and ferritin levels decreased. However, the patient remained respiratory insufficient with progressive renal failure. Synchronising therapy with plasmapheresis, rituximab, and cyclophosphamide was started for treatment of the underlying severe SLE and anakinra was discontinued. Due to subsequent insufficient control of inflammation, therapy was switched to the HLH-2004 protocol. Yet due to several complications, treatment with etoposide was discontinued and anakinra was started again without any clinical response. The patient died within seven weeks after admission.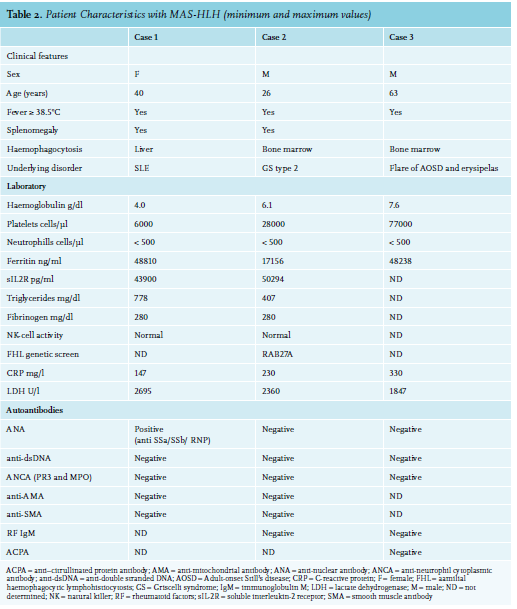
Case 2
A 26-year-old male, originally from Eritrea, presented with a right-sided peripheral facial nerve palsy suspected for Bell’s palsy after a cold and a tick bite. Treatment with prednisone was started. Two days after the first visit, the patient experienced numbness, tingling, and progressive muscle weakness of the lower limbs, resulting in left-sided foot drop. A presumed diagnosis of Guillan-Barre Syndrome was made and intravenous immunoglobulins (IVIG) were started. He showed initial improvement after the first cycle of IVIG therapy. However, 14 days later, he developed a left-sided facial and bilateral abducens nerve palsy, difficulty with speaking and swallowing, symmetric paraparesis of the upper and lower limbs, areflexia, sensibility loss with numbness from the knees down, and also fever. Electromyography and magnetic resonance imaging of the brain were normal. A lumbar tap (2x) was performed without the presence of infectious or malignant cells. Based on clinical course, the diagnosis of polyradiculitis was established and a second IVIG cycle was started. During follow-up, the patient developed respiratory insufficiency, bowel complaints, fever (up to 39.5°C), abnormal liver tests, and decreased haptoglobulin and pancytopenia were observed (table 2). In addition, soluble interleukin-2 receptor (sIL2R), ferritin, and triglycerides levels were raised. Cytotoxic natural killer (NK)-cell function was normal. Renal function and C-reactive protein (CRP) were unremarkable. Blood, urine, spinal fluid, and fecal cultures were negative.
Additional imaging diagnostic tests were normal, except for the splenomegaly (17 cm). Bone marrow biopsy revealed haemophagocytosis and hyperinfiltration of T cells, and the patient fulfilled the criteria for MAS-HLH. Treatment with dexamethasone was started as initial treatment for MAS-HLH. Etoposide was considered at this time, but withheld due to concern of ongoing infection. Although some improvement in pancytopenia was observed, the patient suddenly went into cardiopulmonary arrest and was resuscitated. Despite achieving spontaneous cardiac output, post-anoxic coma developed with a poor prognosis. The patient died approximately seven weeks after his first presentation, despite initiation of etoposide treatment after resuscitation.
Interestingly, post-mortem, a genetic test revealed a homozygote RAB27A mutation. This mutation lead to Griscelli syndrome (GS) type 2, a rare autosomal recessive disorder, which is associated familial/primary HLH. Additional tests on the bone marrow biopsy also revealed an abnormal T-cell population, potentially compatible with T-cell malignancy.
Case 3
A 63-year-old male presented with a two-year history of uveitis anterior, thrombophlebitis, episcleritis, arthritis, auricular perichondritis, pharyngitis and otitis, as well as macular skin lesions and livid macular lesions on the lower extremities. He had developed relapsing and remitting fevers, and neutrophilic dermatosis (Sweet syndrome). PET scan showed flurodeoxyglucose (FDG)-positive consolidations within the lung parenchyma, reactive mediastinal lymphadenopathy, and splenomegaly with diffuse increased uptake in bone marrow with reactive findings on bone marrow biopsy. Repeated infections were ruled out by bronchoalveolar lavage. Intially considered as aspecific inflammatory syndrome, the patient was treated with different disease-modufying anti-rheumatic drugs and prednisone, but tapering of prednisone was followed by disease flares with fever, and eventually skin rash; pancytopenia; raised inflammatory markers (CRP 8.9 mg/dl and erythrocyte sedimentation rate 64 mm/hr), ferritin (up to 2040 ng/ml), and sIL2R (6025 U/ml) levels. He had normal triglycerides, and liver and renal function most compatible with AOSD with concurrent flare of Sweet syndrome. Anakinra was administered, but due to severe injection-site reactions this was switched to canakinumab and with disease control and the patient was dismissed. In the meantime, myelodysplastic syndrome type EB-1 (excess blast-1) was considered based on revised bone marrow examination and he was referred for consideration for an allogeneic stem cell transplant. However, 10 weeks later, the patient presented at the emergency department with bullous skin lesions on the left lower leg, a fever of up to 38.5°C, high CRP (14 mg/dl), normocytic anaemia (93 g/l), leucocytosis (15300 cells/uL), and a normal platelet count. The patient was diagnosed with erysipelas and intravenous antibiotic was initiated. Blood cultures revealed gram-positive bacteria and Pseudomonas species were cultured from the skin lesion. Nevertheless, progressive necrosis developed in the left foot. The patient was transferred to the intensive care unit because of haemodynamic instability. Despite surgery, the patient’s condition deteriorated with a fever of up to 41°C with progressive pancytopenia and abnormal liver tests (AST 1155 U/l). Moreover, ferritin levels were elevated, yet sIL2R and triglycerides were not tested (table 2). Septic shock was assumed as clinical condition, and only by the time of ongoing deterioration, was MAS-HLH considered provoked by a flare of AOSD due to infection. The patient died within 10 days after admission to the hospital. Post-mortem analysis of bone marrow showed an increase in the number of macrophages and focal haemophagocytosis (figure 1), consistent with MAS-HLH.
DISCUSSION
Here we present three adult patients with MAS-HLH with various active underlying diseases, which illustrate the different faces of MAS-HLH. The clinical and laboratory features in all our patients met the requirements for MAS-HLH according to the HLH-2004 guidelines, yet concurred with delayed treatment. Measurent of serum ferritin early in disease course and monitoring for dynamics is inexpensive and may be instrumental in earlier recognition of MAS-HLH.8
Although MAS-HLH is well-recognised in paediatric systemic juvenile idopathic arthritis (sJIA), more data on exceptional presentation of MAS-HLH in other CTD/ inflammatory diseases in adulthood is useful. The current fatal case series described in our study aims to increase awareness of clinical features and outcome of these exceptional presentations of MAS-HLH in SLE, GS syndrome type 2 with possibly lymphoma and infectioninduced AOSD flare.
Macrophage activation syndrome secondary to SLE is rare and the reported prevalence ranges between 0.9 and 4.6%.3,9,10 Recognition of MAS-HLH in SLE patients is challenging, since it can mimic SLE flare, with delay in initiating appropriate treatment. The presence of unexplained fever, pancytopenia, liver dysfunction, jaundice, and high lactate dehydrogenase and ferritin levels unresponsive to immunosuppressive therapy should raise the suspicion for MAS-HLH.10-12 Gavand et al. reported MAS occurring in more than half of their cohort after SLE diagnosis was made with a median delay of 106 months (range 49-177).11 Therefore, the presentation of MAS in our patient at 192 months was relatively late. Indeed, our patient with SLE also presented with these symptoms as first manifestation of MAS-HLH, which was not recognised as such initially, with consequent admission to ICU and delay in treatment. High mortality rates (10-20%) are reported in case series of patients with MAS in studies among SLE patients.3,13
Our second patient, presented with neurological symptoms as manifestation of primary HLH. Central nervous system involvement has been reported in 30% of patients with MAS-HLH. In the current case, HLH was associated with GS type 2, which is a rare and fatal autosomal recessive disorder, due to homozygous pathogenic mutations in the RAB27A gene.5 GS type 2 is associated with impaired pigmentation, primary immunodeficiency due to dysfunction of cytotoxic T cells and NK cells, with subsequent susceptibility to repeated infections and HLH, leading to death without haematopoetic cell transplantation. This patient did not suffer from albinism, which has been described in four patients with the same mutation.14 Also, similar to our patient, late onset presentation of disease has been reported before, yet is very rare,15 and may contribute to late recognition of the syndrome. Our patient had normal NK-cell function, unlike the other patients with the same mutation. An explanation for this may be due to somatic reversion leading to selective outgrowth of particular cell subsets, which has been reported in T-cell subsets in, for example, X-linked severe combined immune deficiency,16 dedicator of cytokinesis 8 deficiency,17 and Wiskott-Aldrich syndrome,18 all autosomal recessive primary immunodeficiencies. Interestingly, post-mortem analysis of the bone marrow also revealed an abnormal T-cell population suggestive of a T-cell malignancy. An association between T-cell malignancies and MAS-HLH has been reported in the literature, in particular, with cutaneous T-cell lymphoma and T/NK-cell lymphomas.19 To the best of our knowledge, there is no correlation reported between GS type 2 and T-cell malignancy.
The third patient had a MAS-HLH associated with infection and a flare of a therapy refractory AOSD all probably due to underlying myelodysplastic syndrome, despite lack of characteristic somatic mutations. Patients with AOSD may experience different life-threatening complications, including MAS, thrombotic thrombocytopenia purpura, pulmonary hypertension, and diffuse alveolar haemorrhage.20 MAS as a complication of AOSD is reported in 10-25% of adult patients. MAS occurs primarily during the onset of AOSD, but can also develop during the course or a flare-up of AOSD.20
Different cytokines are reported to play a role in the pathogenesis of MAS-HLH, however, the involvement of interleukin (IL)-1 seems to play a central role in the pathogenesis, highlighted by the excellent clinical responses to IL-1 blockade in recent studies in sJIA and AOSD-related MAS. Recent observations suggest that initiation of anakinra early in the disease course may improve outcomes of MAS without significant side effects.4,21,23 Furthermore, IL-1 blockade was associated with less mortality and complications (e.g., sepsis, malignancy) compared to the conventional treatment for MAS without masking clinical and biological features of underlying disease.22-25
In addition, a clinical trial is pending on the role of early treatment with IL-1 blockade in patients (both children and adults) with MAS-HLH from the group of Chatman et al. Interestingly, the Food and Drug Administration recently approved the use of emapalumab, an interferon gamma blocking antibody (IFNγ), for the treatment of primary and secondary HLH. Several animal and human studies have reported on elevated IFNγ in MAS-HLH. International phase 2/3 clinical trials are currently ongoing investigating the efficacy, safety, and long-term outcomes in paediatric patients diagnosed with primary HLH and secondary HLH associated with SJIA. Development of more targeted therapeutic options for patients with MAS-HLH may be lifesaving and have fewer side effects and toxicity than the conventional treatment protocol (HLH-2004).26,27
CONCLUSION
MAS-HLH is a potentially fatal complication in a heterogeneous group of underlying diseases, ranging from CTD or infectious disease to haematological conditions. Awareness and early treatment is warranted when diagnosis is suspected.
DISCLOSURE
HL attended advisory boards and received speakers fees from Novartis and Sobi.
REFERENCES