KEYWORDS
MGRS, MGUS, paraproteinaemia-associated kidney disease, plasma cell dyscrasia, monoclonal immunoglobulin, free light chain
INTRODUCTION
A circulating monoclonal protein (M-protein) is present in approximately 3% of people aged 50 years and older and increases to 5% after 70 years of age.1,2 Patients with this circulating M-protein are diagnosed with monoclonal gammopathy of undetermined significance (MGUS) if the M-protein is < 30 g/l and a bone marrow (BM) examination shows < 10% monoclonal plasma cells with absence of end organ damage or other multiple myeloma defining events.2,3 Monoclonal gammopathies can be completely asymptomatic in the case of MGUS, but monoclonal gammopathy is sometimes associated with potentially severe organ damage whereby the criteria for overt multiple myeloma (MM) or other malignant lymphoproliferative disorders such as B-cell non-Hodgkin lymphoma, including Waldenström’s macroglobulinaemia (WM) or chronic lymphocytic leukaemia (CLL), are not met (table 1). 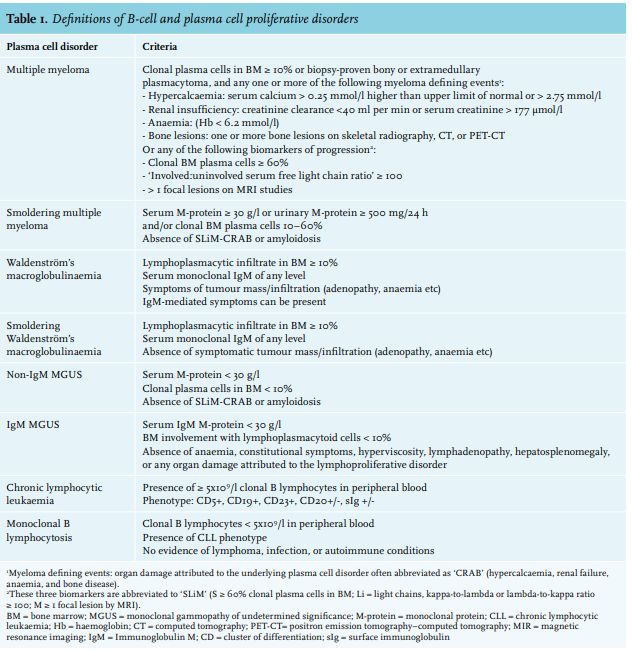
Therefore in 2012, the term monoclonal gammopathy of renal significance (MGRS) was introduced by the International Kidney and Monoclonal Gammopathy Research Group (IKMG) to describe a spectrum of kidney diseases due to M-protein secreted by a small B-cell, lymphoplasmacytic, or plasma cell clone not meeting any current haematologic criteria for specific therapy.1 The term MGRS was recently updated by the IKMG and now includes all B-cell or plasma cell proliferative disorders that produce a nephrotoxic M-protein such as smoldering MM, smoldering WM, and monoclonal B-cell lymphocytosis (MBL). Low-grade B-cell lymphomas and low-grade CLL with associated renal disease are also included in the updated definition of MGRS. M-proteins secreted by this clone can be directly nephrotoxic and also indirectly nephrotoxic by complement activation. In addition to kidney involvement, these small B-cell or plasma cell clones can also involve other organs such as skin and peripheral nerves and therefore, the concept of monoclonal gammopathy of clinical significance (MGCS) was recently introduced.4
The current treatment guidelines do not recommend anti-tumour therapy in patients with MGUS and smoldering myeloma or asymptomatic WM. These patients are usually monitored as they can remain asymptomatic for years and only in the case of developing or impending symptoms can therapy be introduced.3,5 In patients with MGRS, this approach may be inadequate, as the M-protein plays a direct role in the pathogenesis of kidney disease despite the absence of high tumour burden. In these cases, the monoclonal gammopathy is of known significance as kidney disease is present with increased risk of progression to end stage renal disease (ESRD). In addition, cases of recurrence after kidney transplantation have been described.6-15
In this review, we illustrate the spectrum of MGRS with the histopathologic classification, present a renal and haematologic diagnostic workup that may assist in the diagnosis of MGRS, and discuss treatment options for the underlying haematologic disorder to improve renal and patient outcome. Ideally, patients with MGRS should be evaluated by a multidisciplinary team that includes nephrologists, haematologists, and nephropathologists with expertise in these conditions and therefore, we have established a Benelux MGRS Working Group (Belgium, Netherlands, Luxemburg) that can be consulted in cases of suspected or proven MGRS.
EPIDEMIOLOGY OF MONOCLONAL GAMMOPATHY OF RENAL SIGNIFICANCE
Little is known about the epidemiology of most MGRS disorders. Shaik et al. reported a prevalence of MGRS of 6% in individuals diagnosed with MGUS in the United States16 and Steiner et al. reported a prevalence of 1.5% of MGRS in 2935 patients diagnosed with MGUS.17 Regarding specific disease data, amyloid light chain (AL amyloidosis was the cause of ESRD in 1.15% of patients in the Netherlands who started dialysis in 2016.18 Renal biopsy registries show that a diagnosis of light chain deposition disease (LCDD) is made in 0.3-0.5% of all kidney biopsies, with an identified underlying MGUS in approximately 41% of these cases.19,20 All other types of MGRS have unknown incidence and/or prevalence as they are only described in the form of case reports and small case series. Although the recognition of MGRS as a new entity has increased in recent years, under-reporting of this disease still exists.
PATHOPHYSIOLOGY AND HISTOPATHOLOGIC CLASSIFICATION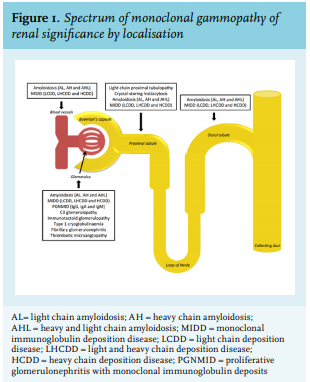
Kidney diseases associated with monoclonal gammopathy are various and the list of recognized disease entities is still expanding. Renal lesions associated with MGRS can be classified by localisation (figure 1) and include glomerular, tubulointerstitial, and vascular patterns of injury, single or in combination.21-23
In addition to a direct toxic effect of M-protein deposits, M-proteins can also have autoantibody activity which results in complement activation and subsequent deposition of complement factors in the kidney (indirect mechanism). The type of kidney injury seems to be dependent on the characteristics of the M-protein.24,25
Light chain (AL), heavy chain (AH), and heavy and light chain (AHL) amyloidosis
Extracellular deposition of amyloid in glomeruli, tubules, and/or vessels is characteristic for renal amyloidosis. In most cases, the M-protein-related amyloidosis is derived from fragments of monoclonal light chains (LCs), which are more often of the λ than κ isotype26, and rarely from fragments of intact immunoglobulin (Ig) or heavy chains only. With light microscopy (LM) amyloid appears as acellular deposits that stain pale eosinophilic on haematoxylin and eosin, periodic acid– Schiff (PAS)-negative or weak, trichrome-blue or gray, and silver-negative (figure 2A). Amyloid is the only MGRS lesion that is Congo red-positive and identified by apple green birefringence under polarised light (figure 2B). With immunofluorescense (IF) microscopy, monotypic staining of the amyloid deposits can be present (figure 2C). With electron microscopy (EM), amyloid deposits in the kidney appear as nonbranching fibrils that are randomly arranged with a thickness between 7 and 14 nm and can be seen within the mesangium, glomerular or tubular basement membranes, interstitium, and vessels.21 AL/AH/AHL amyloidosis is a systemic disease and most patients present with two to four organs involved.27
Monoclonal immunoglobulin deposition disease (MIDD)
MIDD includes three subtypes depending on the composition of the deposits. Light chain deposition disease (LCDD) is the most common subtype, primarily the κ type. The kidneys are almost always affected and extrarenal involvement is common in the heart, liver, and lungs.21 Under LM, a nodular glomerulosclerosis pattern is classic for MIDD and nodular mesangial expansion along with thickening of the glomerular and tubular basement membranes (GBM and TBM) is also present (figure 2D). A variable degree of tubular atrophy, interstitial fibrosis, and inflammation can also be present as nonspecific findings.
With IF, LCDD presents with monotypical linear amorphous LC deposits in the mesangium, and along the glomerular and tubular basement membranes. In light and heavy chain deposition disease (LHCDD) and heavy chain deposition disease (HCDD), linear deposition of monotypic γ, α, or μ HC along the GBM and TBM are seen (figure 2E, F). The granular deposits appear nonfibrillar and electron-dense and are located in the glomerular subendothelium and mesangium, and on the outer aspect of the tubular basement membrane, as observed by EM.21,28,29
Proliferative glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID)
PGNMID is the result of glomerular deposition of monoclonal IgG or rarely, IgA or IgM. LM shows predominantly endocapillary proliferation and/or membranoproliferative glomerulonephritis (MPGN), with or without membranous features. As seen by IF, the IgG deposits are confined to glomeruli and consist of a single light chain isotype and a single heavy chain subtype, most commonly IgG3κ. Positive staining for C3 and C1q indicates activation of complement system. Granular, non-organized deposits, typically in a subendothelial and mesangial distribution are characteristic findings with EM.21,30-32
Monoclonal gammopathy-associated C3 glomerulopathy (C3GP)
C3GP results in kidney dysfunction via an indirect mechanism. In most cases, the mechanism by which the presence of an M-protein, mostly IgG, predisposes to C3GP is not clear. A possible mechanism has been suggested whereby the M-protein acts as an autoantibody to C3 convertase or as an autoantibody to other complementregulating proteins which results in dysregulation of the alternative complement pathway.15,33
C3GP is characterized by glomerular deposition of the complement component C3 fragment at least two orders of intensity stronger than any combination of IgG, IgM, IgA, and C1q. Mesangial proliferative, membranoproliferative, or endocapillary proliferative glomerulonephritis are the usual patterns on LM. Electron-dense deposits located in the mesangium and subendothelium can be seen with EM.34
Immunotactoid glomerulopathy (ITG)
ITG is often monoclonal in nature, in contrast to fibrillary glomerulonephritis (FGN).9 ITG is characterized by either an MPGN or diffuse proliferative pattern as observed with LM. Organized and parallel-oriented microtubular M-protein deposits with either κ or λ light chain restrictions are seen. These microtubular glomerular deposits are mostly composed of IgG, are Congo red-negative, contain distinct hollow centers, and stain for complement (C3) by IF. They are focally arranged in parallel arrays and are usually greater than 30-90 nm in diameter as seen by EM.21,35
Cryoglobulinaemia-associated glomerulonephritis (CGG)
Cryoglobulinaemia (CG) is composed of three types of which type I and type II are associated with monoclonal proteins. Type 1 CG is characterized by precipitation of M-protein when exposed to a temperature lower than 37°C and dissolve once the serum is heated. It can cause CGG or vasculitis. With LM, the characteristic finding of CG is a MPGN with endocapillary proliferation. Numerous intracapillary infiltrating leukocytes and large PAS-positive intraluminal immune deposits (protein hyaline thrombi) can be seen with LM. Intraluminal Ig and deposition of C3, C4, C1q are seen with IF and substructures like microtubules, fibrils, and finger print deposits can be observed with EM.21,31,36 In type II CG, the monoclonal Ig is bound to the constant domain of polyclonal Ig heavy chains forming a complex. The most common cause of type II CG is hepatitis C infection. Approximately 10-30% of cases are not related to hepatitis C infection but to B-cell lymphoproliferative disorders.37
Paraprotein-associated fibrillary glomerulonephritis (FGN)
Although FGN is often polyclonal in nature, it can also be associated with organized glomerular M-protein deposits which have a fibrillary amyloid-like appearance as observed with EM. An MPGN pattern is commonly seen with LM. Unlike amyloid, lesions are Congo red-negative.38 IF staining for IgG (mostly IgG4, sometimes IgG1) and C3 is common in these fibrils, which have a diameter ranging from 16 to 24 nm, as observed with EM.35
Monoclonal gammopathy-associated thrombotic microangiopathy (TMA)
TMA associated with monoclonal gammopathy is a relatively new entity with few cases described in the literature. TMA with no other cause than gammopathy was first described in three cases that were part of a large cohort of patients with WM and a number of single case reports.39 The causal relation between the gammopathy and TMA was further supported by an unexpected high occurrence of monoclonal gammopathy in patients with TMA reported by Ravindran et al.,40 with an incidence of 21% in patients aged 50 and older. How monoclonal gammopathy induces TMA is not fully understood and remains to be elucidated. Kidney injury in TMA is characterised by thrombus in the glomerular capillaries, swelling of the endothelium, mesangiolysis with microaneurysms, and double-contour formation of the glomerular capillary walls. Acute tubular injury with differing degrees of tubulointerstitial scarring and thrombus in the arteries and arterioles can also be seen.40
Light chain proximal tubulopathy (LCPT)
LCPT is characterised by proximal tubular dysfunction secondary to cytoplasmatic inclusions of LCs as crystals within the endolysosomal compartment of proximal tubular cells. The accumulated LCs are almost always of the κ class and are rod- or rhomboid-shaped hypereosinophilic and PAS-negative under LM (figure 2G, H). For the visualization of the intracytoplasmic crystalline inclusions of κ LC in proximal tubular cells by IF, pronase digestion might be necessary.41 The electron-dense intracytoplasmic inclusions can be seen with EM (figure 2I). Some inclusions appear to be membrane-bound within the endosomes or lie free in the cytosol.42 Recently, it was shown that LCPT without crystal formation is actually more common than LCPT with crystal formation and is usually associated with accumulation of λ LCs. LCPT with crystal formation seems to be more frequent in younger patients and is associated with Fanconi’s syndrome (FS) and a worse kidney function compared with noncrystalline LCPT, as reported in one study.42 However, little is known about the clinical significance of crystalline vs noncrystalline deposits because of their rarity.43 It could be argued whether noncrystalline LCPT is pathologic, given the physiologic reabsorption of LCs occurring in the proximal tubule.
Crystal-storing histiocytosis (CSH)
In CSH, the M-protein crystals accumulate within the lysosomes of histiocytes accumulated in the interstitium. LM shows that the crystal-containing histiocytes are accompanied by interstitial fibrosis and tubular atrophy and IF demonstrates that the accumulated crystals are almost always LCs of the κ class. Diagnosis of CSH can be challenging since the crystalline inclusions cannot always be identifyed by IF and pronase-digested tissue or the immunoperoxidase method may be required.41,44 CSH can occur simultaneously with LCPT. If this is the case, organized cytoplasmic inclusions of needle- or oval-shaped crystals in proximal tubular cells are seen with EM.21,45
DIAGNOSIS OF MGRS
When should MGRS be considered? 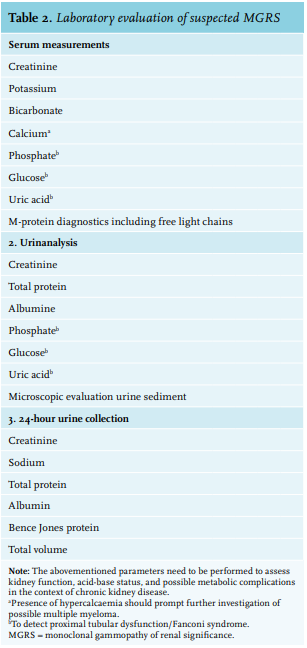
Clinical manifestations depend on the affected segment of the nephron. Patients present with a progressive decline in kidney function, microscopic haematuria, proteinuria (ranging from subnephrotic to overt nephrotic syndrome), and/or proximal tubular dysfunction.21 The proximal tubular dysfunction can result in FS which is characterised by normoglycaemic glycosuria, hyperphosphaturia resulting in hypophosphataemia, proteinuria, aminoaciduria, hyperuricosuria, and urinary wasting of bicarbonate. Incomplete FS has also been reported.31
Therefore, MGRS should be considered in every patient with renal manifestations combined with an M-protein, especially when classical cardiovascular risk factors or diseases, such as diabetes mellitus, hypertension, and atherosclerosis are absent or well controlled. MGRS should also be considerd in patients presenting with C3GP and TMA, which are known to have a strong association with M-protein and kidney disease, for example, in elderly patients. Although MGRS is commonly seen in patients of 50 years and older14,46, it has also been reported in younger patients.9-11,14,47
Extrarenal manifestations are common in AL amyloidosis and MIDD, with the heart and liver often affected, which could serve as a clinical clue. In type 1 CG, the skin is the most affected organ.48 However, these patients are difficult to recognize and diagnosis often requires extensive consultation between nephrologist and haematologist.
In the diagnosis of MGRS there are two important objectives: 1) one should try to identify the circulating M-protein and 2) one should try to establish a causal relationship between the M-protein and the observed renal abnormality. Therefore, kidney biopsy is essential and should be performed in all cases of suspected MGRS. Laboratory evaluation in patients suspected of having GRS is summarised in table 2.
Monoclonal protein testing
Typically, MGRS exhibits low levels of circulating M-protein reflecting the small size of the underlying B-cell or plasma cell clone.21 With detection limits of 500-2000 mg/l, serum protein electrophoresis (SPE) has insufficient sensitivity to detect low levels of M-protein, particularly free light chains (FLCs) because of their lower half-life compared to intact Ig (table 3).49-51 Kyle et al. found that an M-protein cannot be detected by routine SPE in > 50% of the patients with AL amyloidosis.52 Serum immunofixation electrophoresis (IFE) is approximately 10-fold more sensitive than SPE, yet urine IFE and/or serum free light chain assay (sFLC assay) are usually necessary to detect an M-protein.49
An sFLC assay utilizes antibodies directed against epitopes that are exposed in “free” LCs but hidden in intact Ig molecules. This assay provides a quantitative measurement of both κ and λ FLCs with a sensitivity of < 5 mg/l, and calculation of a κ/λ ratio can detect unbalanced LC synthesis as a surrogate marker for a monoclonal gammopathy.49,50 Studies evaluating the diagnostic performance of this test have documented detection of abnormal κ/λ ratios in 100% of patients with LC MM49,52, in 76-98% of patients with AL amyloidosis52-55, and in 92-100% of patients with LCDD.56-58
For more than 10 years, sFLC quantification has been performed by an immunonephelometric assay based on polyclonal antibodies (Freelite®, The Binding Site, Birmingham, UK). In recent years, novel assays based on monoclonal antibodies have entered clinical practice.51,59,60 The Freelite FLC assay and one of the novel assays (N Latex FLC assay®, Siemens Healthcare Diagnostic Products GmbH, Marburg, Germany) seem to have similar diagnostic performance, though current data indicate that they are not interchangeable, especially in monitoring response to therapy.59. 61,62
Reference intervals for the Freelite serum FLC were defined by Katzmann et al. (normal range κ: 3.3-19.4 mg/l; λ: 5.7-26.3 mg/l; κ/λ ratio: 0.26-1.65).56 Since FLCs are filtered by the glomerulus and metabolised in the proximal tubules, caution is required when interpreting this assay in the context of renal impairment. As glomerular filtration rate (GFR) significantly declines, the removal of FLCs by reticuloendothelial pinocytosis becomes more important resulting in a prolonged FLC half-life and polyclonal increase in both κ and λ FLCs.63 In patients with normal GFR, the increased physiological production of polyclonal κ LCs (molecular weight (MW) 22.5 kDa) is masked by the more rapid clearance of this monomeric LC compared to larger dimeric λ LCs (MW 45-50 kDa).64 As the differential ability to clear κ and λ LCs by the kidney is lost, the κ/λ ratio can slightly increase and, for this reason, adaptation of the normal range to 0.37-3.17 increases the reliability of the Freelite FLC assay in patients with renal impairment.65 In the BeNeLux region, most clinical laboratories make use of the Freelite FLC assay, which has an adjusted FLC ratio reference range of 0.37-3.17. In terms of M-protein diagnostics, a patient with MGRS and renal impairment would reach complete response (CR) in case of negative IFE in serum and urine. Provided that CR criteria are met and clonal cells in bone marrow are absent, the patient reaches serum CR when the adjusted FLC ratio is normal.
On the contrary, when using the N Latex FLC assay, there is no need for a separate renal reference range since the κ/λ ratio in patients with kidney disease does not differ from the normal values in healthy controls.66 Adaptation of the κ/λ ratio in the first assay and the unadapted κ/λ ratio in the second assay, illustrate the heterogeneity that exists in FLC measurement. Therefore, follow-up of FLC measurements should be performed using the same assay and the same platform.62
International guidelines recommend using an sFLC assay along with SPE and IF as an initial screening panel for monoclonal gammopathies, because of the incremental sensitivity and potential limitations of urinary assessment in this setting.67,68 Since FLCs only appear in the urine when the proximal tubular reabsorption capacity is overwhelmed, low-level serum FLCs will not be detected by urine tests.49 Furthermore, urine analysis can be unreliable due to errors in carrying out a correct 24-hour urine collection, or due to inaccurate interpretation of EP results in concentrated urine samples or nephrotic-range proteinuria.49 On the other hand, several studies have proven the necessity to perform both serum and urine IF for optimal sensitivity in AL amyloidosis and LCDD.54 In addition, since 24-hour urine collection is necessary to calculate the GFR more precisely and measure the amount of proteinuria, urine IF and EP can be performed on this sample. 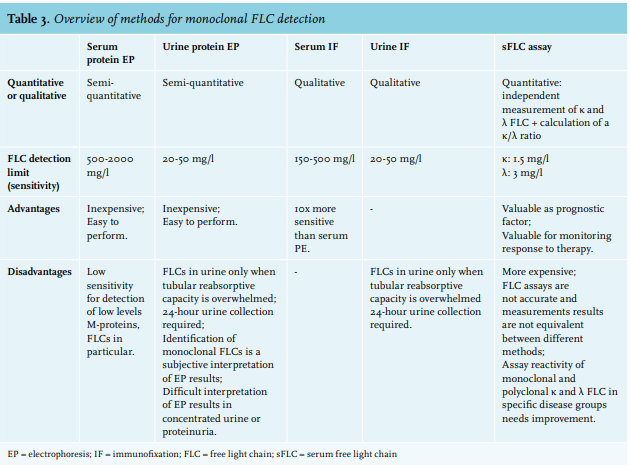
Kidney biopsy
A kidney biopsy is crucial for the diagnosis of MGRS to determine whether the M-protein is an innocent bystander or the cause of the renal disease. Detailed IF, immunohistochemistry (IHC), and EM studies to identify deposit composition and ultrastructural organisation pattern should be applied. In this respect, it is important to emphasize that EM should be part of the standard work up, since FGN, ITG, type 1 CG and crystalline LCPT can only be diagnosed with EM. An exception can be made for patients with suspected AL amyloidosis, in whom other tissue specimens (e.g., abdominal fat) can be sufficient for diagnosis if clinical criteria for renal involvement are met.69
In selected cases, more sophisticated techniques such as immunogold EM or proteomics via laser microdissection and by mass spectrometry (LMD/MS) are required to characterise the component proteins of dense deposits.70,71 LMD/MS is not only considered the gold standard for accurate typing of amyloidosis, it has also proven to be extremely useful for correct diagnosis and understanding of other MGRS.72,73 The use of these advanced techniques may require sending the biopsy sample to a specialised centre and is therefore usually reserved for equivocal results or difficult cases.
Bone marrow
In order to identify the underlying B-cell or plasma cell clone, which is essential to determine treatment strategy, a detailed haematologic evaluation should be performed in all cases.21,37 Overt MM or WM should be excluded, and BM aspirate and biopsy generally show only a small increase in plasma cells or B-cells (by definition < 10%) without morphologic abnormalities.21,37,74 A Congo red stain performed on the BM biopsy could demonstrate amyloidosis which is present in approximately 60% of AL amyloidosis cases.75 IHC and/or flow cytometry is mandatory to establish clonality and to make the connection between the circulating M-protein and the presence of this M-protein in the kidney.21,37
TREATMENT OF MGRS
The aim of the treatment in MGRS is to preserve or improve organ function by targeting the B-cell or plasma cell clone that is responsible for production of M-protein and organ damage. Achieving complete haematologic response will lead to better organ response and thereby prevent progression of organ damage.37 Current evidence strongly supports the strategy of clone-directed therapy.
Except for amyloidosis, very few data regarding treatment options exists for the other MGRS (sub)types and there are currently no evidence-based recommendations. Based on literature concerning MM and AL amyloidosis, it can be assumed that when treating MGRS, the best results are achieved by targeting the underlying MGUS clone and aiming for a deep and prolonged haematologic response. Treatment depends on the isotype of the underlying clone in the BM (IgG, IgA, or LCs only versus IgM clone), the renal metabolism and potential renal toxicity of the therapy, and presence of neuropathy in the patient.37
Plasma cell dyscrasia with IgG, IgA, or LCs only
In case of an IgG, IgA, or LC-only-producing plasma cell clone (non-IgM MGUS), therapy directed at eradication of the plama cell clone with anti-myeloma agents should be considered to preserve kidney function. The most important drug in the treatment of MGRS associated with a plasma cell clone is the proteasome inhibitor bortezomib. Bortezomib has a nonrenal metabolism and is usually given in combination with dexamethasone. Other proteasome inhibitors are currently available, but bortezomib has the most robust data in the treatment of MGRS. In MM patients with renal impairment, bortezomib-containing regimens have demonstrated rapid reduction of tumour load and improved kidney function. In addition, in a study with 27 dialysis patients with MM, bortezomib was used as induction therapy and showed a higher overall response rate compared to induction therapy with conventional chemotherapy, and reduced the transplant-related mortality.76,77 Bortezomib has been demonstrated to be highly effective drug in AL amyloidosis and seems to be the most effective agent in MIDD as high rates of CR/very good partial response (VGPR) have been described after treatment with bortezomib-based regimens and melphalan-conditioned autologous stem cell transplantation (ASCT).12,77-79 Furthermore, integration of bortezomib both before and after ASCT overcomes the negative impact of renal failure in AL amyloidosis.80-81
For an optimal treatment strategy, it is important to determine whether patients are eligible for ASCT since this therapy can deepen and prolong the duration of remission. Similar to MM, ASCT is preceded by high-dose therapy with melphalan. The melphalan dose is normally reduced in cases of GFR of < 40 ml/min. Induction therapy prior to ASCT can be omitted in cases of a small clone, but proves to be beneficial for patients with a poor performance status due to MGRS. The treatment-related mortality of ASCT is < 1%.
Determination of eligibility for ASCT differs across countries and institutions. However, the decision to undergo ASCT should be made on a case-by-case basis by reviewing the risk-benefit assessment, and the needs and wishes of the patient. In general, patients younger than 70 years of age, with sufficient cardiac function (ejection fraction > 45%), sufficient pulmonary function, a systolic blood pressure of > 90 mmHg, World Health Organisation performance score < 2, and New York Heart Association scores I-II, are eligible for ASCT. Interestingly, kidney disease is not a contraindication to ASCT, although it is crucial that the renal function is stable. Kidney disease has no adverse effects on the quality of stem cell collection or their engraftment and ASCT is possible in dialysis patients.82-88 However, the procedure is associated with increased risk of transplant-related mortality for patients with kidney disease compared to those with normal kidney function.86 Patients who do not meet the abovementioned criteria and who also have an active infection are not eligible for ASCT. Overall survival is strongly dependent on the haematologic response according to most AL amyloidosis data.86,87 Since MGRS is more similar to MGUS with regard to few plasma cells in the bone marrow, it would be fitting to treat patients who are ineligible for ASCT, with six to eight courses of bortezomib in combination with dexamethasone and cyclophosphamide, or bortezomib plus melphalan-prednisone. Treatment with immunomodulatory drugs (IMiD) such as thalidomide and lenalidomide can be used as an alternative, and in a relapse setting, pomalidomide can be used. Unlike lenalidomide, thalidomide and pomalidomide are not excreted by the kidneys and dose modifications are therefore not needed.89,90 However, lenalidomide has been shown to be more effective than thalidomide and is thus the preferred alternative to bortezomib.
B-cell clone with IgM M-protein
Since IgM MGUS is rare, there is little evidence supporting the choice of treatment in MGRS-related to IgM M-protein. When the underlying BM clone is an IgM M-protein-producing and CD20-expressing B-cell or lymphoplasmacytic clone, rituximab-based therapy is the first choice of treatment. Rituximab can be combined with dexamethasone and cyclophosphamide or bendamustine in MGRS associated with IgM-MGUS. Rituximab can be safely administered without dose modification in patients with decreased kidney function.91 Approximately 60% of cyclophosphamide is eliminated through the kidneys. Studies describe an increase of cyclophosphamide exposure in patients with kidney disease, however, dose adjustments in these patients remain variable in the literature.90,92 Bendamustine does not appear to alter the pharmacokinetics in moderate renal impairment but limited data suggest toxicity increases in patients with GFR < 40 ml/min.93 The benefit of ASCT in these patients has not been demonstrated but since a deep haematologic response is most likely beneficial, this option should also be considered.
The optimal treatment strategy of an IgM-related MGRS disorder should be constructed with the consultation of experts on MGRS, for example, the multidisciplinary Benelux MGRS Working Group, with experience in the use of these drugs.
Supportive care
In all patients, hypertension and/or proteinuria should be treated, preferably with renin-angiotensin system (RAS) inhibitors combined with salt restriction. Diuretics can have an extra antihypertensive and/or antiproteinuric effect. In case of AL amyloidosis, treatment with RAS blockade should be prescribed with caution as these patients have a tendency towards hypotension. In FS, prevention of osteamalacia with bicarbonate, phosphate, and vitamin D supplementation should be considered.22
Response to therapy
In MGRS, assessment of the haematologic response to treatment is crucial because the renal response is dependent on the haematologic response. For example, renal response rate in AL amyloidosis was significantly higher in patients with > 90% suppression of the nephrotoxic M-protein.87 In MIDD, achieving haematologic complete response showed similar benefits for the kidney.79 In AL amyloidosis, measurement of the sFLC is an essential tool for the assessment of a haematologic response. The response criteria used in AL amyloidosis are CR, VGPR, partial response (PR), and no response (NR)94, and it seems logical to use these same response criteria in the other MGRS disorders. The use of sFLC assay for response has been suggested for all MGRS involving LCs only.22 In cases of an undetectable or difficult to measure M-protein, the haematologic response can be assessed with repeated bone marrow examinations using sensitive multicolor flow cytometry. Sometimes, the GFR and proteinuria can be the only parameters used to assess disease activity.94 It is important to note that the renal response is usually delayed. In one study, a minimum duration of 12 months of haematologic response was needed until onset of renal response was seen in patients with AL amyloidosis.95
CONCLUSION
In summary, MGRS consists of a variety of kidney diseases that are caused by the production of an M-protein resulting in kidney deposits and/or autoimmune activity. Histopathologic identification and classification are crucial for determining the optimal treatment for MGRS. For the diagnosis of MGRS, it is important to identify the circulating M-protein and to establish a causal relationship between the M-protein and the renal damage. A kidney biopsy with detailed examination by LM, IHC, IF, and EM is needed in the diagnosis and aids in characterising the MGRS lesion.
Clone-directed therapy is currently the most effective treatment strategy. Choice of therapy will depend on type of clone, eligibility for ASCT, side effects, and scarce literature support. Preferably, patients should be managed by a multidisciplinary team consisting of nephrologists, haematologists, and nephropathologists, with expertise in MGRS for optimal care. In cases of suspected or proven MGRS, the Benelux MGRS Working Group can be consulted during diagnosis and treatment of possible MGRS patients.
DISCLOSURES
All authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES