
KEYWORDS
Creutzfeldt-Jakob disease, Sporadic, Wernicke encephalopathy
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) is a neurodegenerative disorder. It is a transmissible prion disease whereby incorrectly folded prion proteins are formed.1,2 There are several forms of the disease, depending on the prion subtype. Recognition of the disease is exceedingly difficult.3,4 We describe a 65-year-old man with the sporadic form of CJD mimicking Wernicke encephalopathy.
CASE REPORT
A 65-year-old man was referred to the department of internal medicine by the general practitioner due to complaints of progressive cognitive impairment, which consisted of memory loss, loss of organisation and disorientation. He also had a drunken man’s gait and double vision. These complaints had been present for approximately four weeks.
The medical history revealed a myocardial infarction and chronic obstructive pulmonary disease. The patient was not on any medication. He drank 6 units of beer a day. Previously, his alcohol consumption had been considerably higher.
The general physical examination showed no abnormalities. On neurological examination he had nystagmus, a paresis of the right lateral rectus muscle of the eye and a wide gait. Additional laboratory tests and a CAT scan of the brain showed no abnormalities. The most obvious diagnosis was acute Wernicke encephalopathy. We treated the patient immediately with thiamine 500 mg three times a day. After two days the paresis of the lateral rectus muscle showed some improvement. We sent the patient home with the advice to take 100 mg thiamine a day. His wife was involved in the care and checked the correct intake of medications.
However, six weeks later the patient’s cognitive function had regressed and he was totally dependent in his activities of daily living. He also had a dysarthric speech, nystagmus, ataxia and myoclonus of several muscle groups. The diagnosis of rapidly progressive dementia was made, with the differential diagnosis limbic encephalitis, malignancy or Creutzfeldt Jakob disease (CJD). Meanwhile, the results of the vitamin B1 level proved to be normal, whereupon the diagnosis of acute Wernicke encephalopathy was rejected. Tests for paraneoplastic antibodies were negative and the level of protein 14-3-3 in cerebral spinal fluid (CSF) was still pending. An MRI scan (with diffusion weighted imaging) of the brain gave no new insights. The EEG showed a diffuse encephalopathic pattern with delta waves parieto-temporally and sharp-slow waves without triphasic complexes, which made a neurodegenerative condition more likely.
During the following days there was a rapid progression of the symptoms, as well as hypertonia in the arms and legs. The patient could barely speak. The diagnosis of CJD was most likely and the infaust prognosis was shared with the family members. We contacted the physicians of the Registration Centre of Prion Diseases of the Department of Epidemiology, ErasmusMC Rotterdam, who informed the public health authorities. They contacted the family members to inform them about the importance of performing an autopsy.
The patient died the following day, two months after the first symptoms emerged. During the autopsy of the brain, deposits of prion proteins were found, both synaptic as well as perineural and perivacuolar (figure 1). The 14-3-3 protein was detected in CSF. Gene analysis was negative for the familial form of CJD. These findings confirmed the sporadic form of Creutzfeldt-Jakob disease.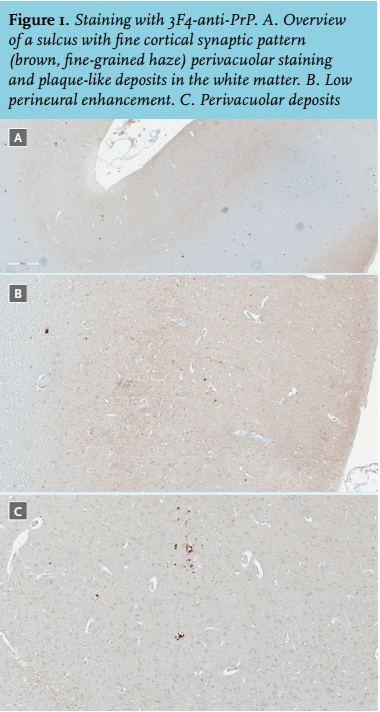
Pathogenesis and clinic
CJD is a communicable and fatal neurodegenerative disease caused by accumulation of natural misfolded proteins (prion proteins) in the body, which will deform the central nervous system.1,5 Neural loss, astrocyte proliferation, spongiform changes and deposition of proteins in the brain tissue will occur.
CJD is divided into multiple forms, depending on the cause and clinical pathological profile (table 1). The sporadic form occurs in 85% of all cases of CJD, with an annual worldwide incidence of 1-2 persons/million. It occurs particularly in people between the ages of 50-70 years.4 The cause is unclear, but there may be a spontaneous change of the structure of prion protein or a spontaneous mutation in the PRNP gene encoding.
Individuals can present with the same symptoms in a different sequence, which makes the disease difficult to recognize in the early stages.3,6,7 It is not uncommon that an alternative diagnosis is suspected, which was the case with our patient.
There are always signs of a rapidly progressive dementia, developing over a period of several weeks or months, in combination with other symptoms such as cerebellar ataxia, loss of vision and myoclonus. However, the disease can also present with isolated cerebellar ataxia or an isolated loss of vision. Characteristically the symptoms are rapidly progressive. The patient can deteriorate in a matter of days. Classical symptoms at the end stage of the disease are akinetic mutism and myoclonus, which were present in our patient. The median duration of the disease is four months.3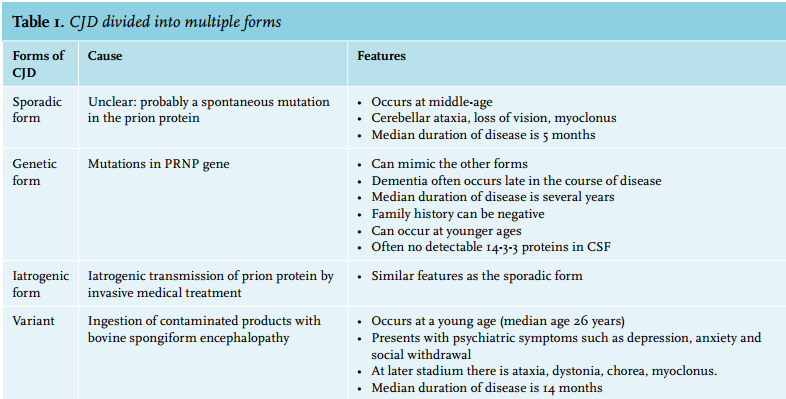
Diagnosis and treatment
As previously described,8 often the diagnosis cannot be made with certainty during the patient’s life. To diagnose CJD with certainty examination of (post-mortem) brain tissue showing the presence of prion proteins in the brain tissues is required. In exceptional cases a brain biopsy is performed while the patient is still alive if there are clear MRI changes, which can be biopsied. The sensitivity of the detection of 14-3-3 protein in CSF (a nonspecific marker of rapid neuronal loss) is 94-97% and the specificity is 84-87%.9,10 The sensitivity of detection of periodic sharp wave complexes in the EEG is 66% with a specificity of 74%. According to the diagnostic criteria, six weeks after presentation the probable diagnosis ‘sporadic CJD’ could have been made in our patient due to rapid progressive dementia, visual and cerebellar problems and myoclonus (table 2).11
To date, there is no curative treatment for this disease and the treatment is only supportive.
Pitfalls
CJD can mimic acute Wernicke encephalopathy with the classical triad of cognitive impairment, nystagmus and ataxia caused by a vitamin B1 deficiency, which is typically seen in malnourished patients, for example in alcohol abusers.12 In our patient this triad was present. Combined with the alcohol abuse, the diagnosis was plausible. However, the non-significant improvement of the paresis of the lateral rectus muscle after supplementation with thiamine did not seem to support this diagnosis.
With the onset of new symptoms a few weeks later, the diagnosis of CJD became clear, enabling us to inform the family members of the situation.
CONCLUSION
The rarity of CJD and the diverse clinical symptoms make recognition of the disease extremely difficult. Rapid recognition of the disease is important so that the patient and relatives can be informed as early as possible during the course of the disease. It is essential to inform the relatives fully to create awareness that deterioration of their loved one can be expected within a period of weeks without possible treatment. Also of importance for the relatives is the information about whether the CJD is the familial form. Because the diagnosis can only be made with the examination of (post-mortem) brain tissue, family members should be extensively counselled about the importance of performing an autopsy.
ACKNOWLEDGEMENTS
We thank Wim van der Hecke, pathologist at the University Medical Centre Utrecht, the Netherlands for providing figure 1 and Alice Moonen, palliative care nurse at the Erasmus Medical Centre Rotterdam for critically reviewing the manuscript regarding English grammar and spelling. Furthermore we would like to thank the family of our patient for granting informed consent to publish this case report.
DISCLOSURES
The authors declare no conflicts of interest. There is no funding or financial support.
REFERENCES