
KEYWORDS
Arrhythmia, hypokalaemia, intake, potassium, redistribution, diuretics
INTRODUCTION
Hypokalaemia is a common clinical problem. It is defined as a serum potassium concentration lower than 3.5 mmol/l. Low serum potassium will increase the transmembrane gradient and therefore hyperpolarise the cell, which impairs the ability of the muscle to depolarise and contract. This can lead to severe disturbances in cardiac, neurological and muscle function.1
We report a case of extreme hypokalaemia leading to ventricular fibrillation. The profound hypokalaemia was likely aggravated by a very deficient diet and use of a thiazide diuretic; our patient never reported any symptoms. An acute intracellular shift of potassium aggravated the pre-existing hypokalaemia. To our knowledge, this is the lowest potassium level reported in literature.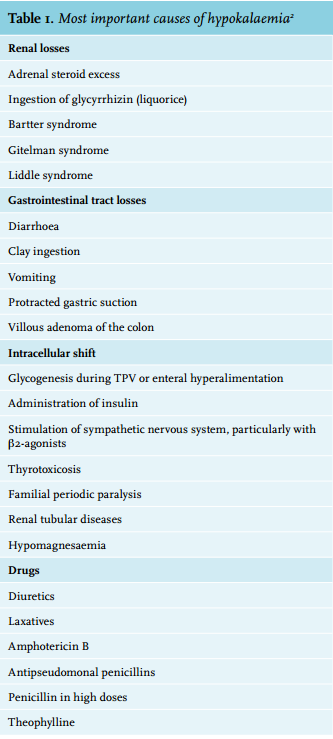
CASE REPORT
A 45-year-old Caucasian woman was found unconscious at home. Upon arrival of the paramedics, the patient was unresponsive to stimuli but otherwise stable. There were no indications of a suicide attempt. In hospital she developed ventricular fibrillation; countershock therapy was followed by a pulseless rhythm. After 26 minutes of cardiopulmonary resuscitation and 6 mg of epinephrine, spontaneous circulation returned.
The electrocardiogram showed diffuse myocardial ischaemia with normal waveform and interval durations. CT scans of the cerebrum and thorax showed no abnormalities. Mild hypovolaemia was suspected following the presence of hypotension (90/60 mmHg) and sinus tachycardia, for which fluid replacement and norepinephrine were started. Blood sampling (table 1) revealed severe acidosis (pH 7.02) with lactic acid > 20.0 mmol/l and extreme hypokalaemia (0.9 mmol/l). A random urine sample showed low potassium and normal sodium concentrations; however, interpretation was difficult due to the very low creatinine level. Toxicology screening and screening for laxatives were negative.
Her history included hypertension, anxiety disorder and heroin abuse. She never reported any muscle weakness, vomiting or diarrhoea. Her dietary pattern was very deficient, with a high consumption of Red Bull energy drinks. She used metoprolol, valsartan, hydrochlorothiazide, clomipramine and methadone. The last routine laboratory test, including potassium, was performed one year earlier and showed no abnormalities.
She was admitted to the ICU and we started intravenous potassium supplementation at a constant rate of 20 mmol/ hour. In the following hours, the serum potassium level increased very slowly; a total amount of 300 mmol potassium was administered to achieve a nearly normal value of 3.2 mmol/l. Because of hypophosphataemia, phosphate supplementation was given. Lactic acid levels decreased quickly and no cardiac events reoccurred. However, her neurological status was poor due to postanoxic injury and the somatosensory evoked potential test showed absent cortical responses. After withdrawal of ventilatory support, death was pronounced. Autopsy was performed; other than chronic thyroiditis there were no relevant findings. Extreme hypokalaemia was listed as the cause of death.
DISCUSSION
The aetiology of hypokalaemia is often apparent from the history or physical examination. Most cases result from unreplenished urinary or gastrointestinal losses, sometimes induced or aggravated by medication. Decreased intake alone rarely causes significant hypokalaemia. Serum potassium levels can be transiently decreased by the entry of potassium into cells (table 2).
In humans, the total body potassium is about 3500 mmol. On average, a 0.3 mmol/l decrease in serum potassium corresponds to a 100 mmol deficit in total body potassium.3
With this formula we estimated the total body potassium deficit in our patient to be at least 800 mmol. However, our patient only needed 300 mmol of potassium to achieve a nearly normal serum potassium concentration. This indicates that, alongside an absolute potassium deficit, a significant part of the decrease in serum potassium was caused by a shift to the intracellular space.
The cause of hypokalaemia is often apparent from the history or physical examination. Our patient’s history was remarkable for medication, a deficient diet and excessive consumption of energy drinks. There were no signs of intoxication or gastrointestinal losses. Chronic thyroiditis was found during autopsy. Thyroiditis may cause periodic paralysis when accompanied by thyrotoxicosis.4 In our patient this is an unlikely cause given the measurement of TSH and fT4 (table 1).
Diuretics are an important cause of renal potassium loss; they stimulate the renin-angiotensin-aldosterone pathway, increase distal tubule flow, increase the release of vasopressin and generate alkalosis, all of which enhance potassium secretion.5 Valsartan, on the other hand, elevates potassium levels by decreasing aldosterone. When hydrochlorothiazide and valsartan are combined, the different effects on serum potassium balance each other in many patients.6 Interpretation of the urine sample was difficult. The urine was very dilute, as demonstrated by the low creatinine concentration. Some authors have argued that a urinary potassium-to-creatinine ratio of 2.5 is the cut-off between renal potassium loss and an adequate renal response.7 In this case the urinary potassium-to-creatinine ratio was 12, indicating renal potassium loss. However, a low potassium concentration with a normal sodium concentration is more suggestive of renal potassium sparing. There was no time for a 24-hour urine collection. We could not calculate the transtubular potassium gradient in retrospect since serum and urine osmolarity were not determined. Hence, we could neither prove nor disprove renal potassium wasting.
Metoprolol, clomipramine and methadone do not affect the potassium concentration. However, methadone might be interesting in this case since it is a potent blocker of IKr potassium channels found in myocytes. In susceptible individuals this leads to QT-complex prolongation, a condition that predisposes to fatal arrhythmias.8 The electrocardiogram of our patient showed normal interval durations. Clenbuterol adulteration of heroin should also be considered a cause of hypokalaemia in drug users;9 our patient had not used heroin for several years and toxicology screening was negative.
By the time we took the first blood sample from our patient, she had received several doses of epinephrine. Catecholamines promote a transient shift of potassium into cells by stimulating membrane Na-K-ATPase activity.10 A proposed physiological role for this effect of increased beta-2 adrenergic activity is to moderate the acute hyperkalaemia of exercise.11
Energy drinks such as Red Bull contain high amounts of caffeine. Caffeine and its breakdown product theophylline will not contribute to sustained hypokalaemia, but can induce an inward cellular shift of potassium by antagonism of the adenosine A2 receptor, resulting in the release of catecholamines.12 Earlier it was believed that increased Na-K-ATPase activity through inhibition of phosphodiesterase was the main mechanism of action, but this requires much more caffeine than consumed through food and drinks.13
Potassium is commonly found in a variety of unrefined foods such as meat, fish, vegetables and fruits. The World Health Organization suggests a potassium intake of 90 mEq/day. Food processing reduces the amount of potassium in food products, and a diet high in processed foods and low in fresh fruits and vegetables is often lacking in potassium.14
A striking fact in this case is the co-existence of extreme hypokalaemia and severe metabolic acidosis, a peculiar situation because acidosis will elevate the serum potassium concentration. When acidosis is present, cells absorb hydrogen ions to raise the serum pH and release potassium in exchange.15 This implies that with a normal serum pH, as probably was the situation before cardiac arrest, serum potassium concentrations might have been even lower than 0.9 mmol/l. On the other hand, it is likely that elevated catecholamine levels during resuscitation worsened the pre-existing hypokalaemia. This makes it difficult to reason about the situation before cardiac arrest. No sodium bicarbonate was given to correct the lactic acidosis.
We did not further investigate the hypophosphataemia in our patient. We suggest that an inward cellular shift played an important role. Total body stores might have been deplete as well; possible contributors are a deficient diet or urinary losses caused by diuretic use and metabolic acidosis.
To our knowledge, this is the lowest potassium level ever reported in the literature. The combination of reduced dietary potassium intake and use of a thiazide diuretic most likely caused the profound hypokalaemia. An acute intracellular shift of potassium due to epinephrine and perhaps also the cathecholamines in Red Bull further decreased the serum potassium concentration. Longer-lasting hypokalaemia might be asymptomatic but when combined with even minor triggers of acute hypokalaemia, fatal arrhythmias can suddenly occur. Patients on diuretic treatment, patients with suspected malnutrition or chronic gastrointestinal losses require regular monitoring of electrolytes.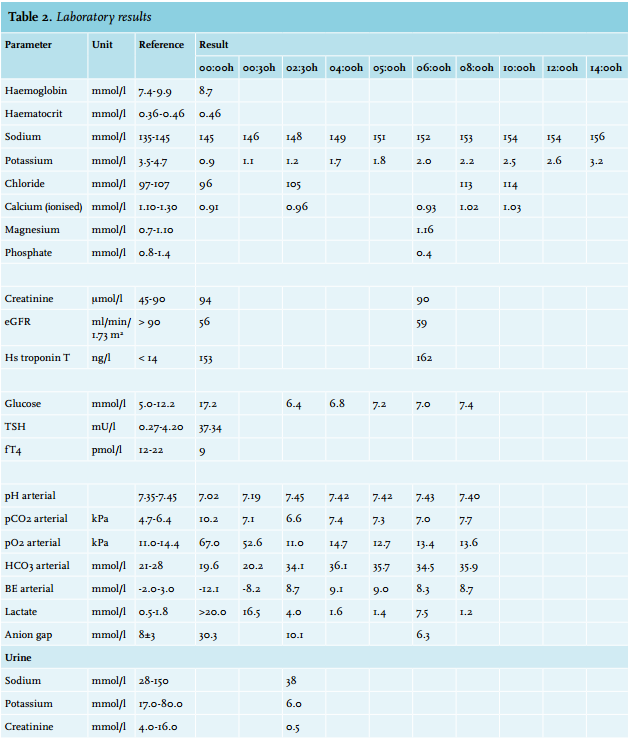
DISCLOSURES
The authors declare no conflicts of interest regarding the publication of this paper. No funding or financial support was received.
REFERENCES