INTRODUCTION
Susac syndrome is a rare disease, named after John Susac (1940-2012) who in 1979 described two female patients with the classical triad of subacute encephalopathy, branch retinal artery occlusion (BRAO), and sensorineural hearing impairment. 1 To date, around 300 cases have been reported in the medical literature worldwide. 2 The course of the disease can be variable, from an initial good response to treatment with full recovery, to refractory cases with persistent, severe encephalopathy, visual and/or hearing loss with major implications on quality of life.
Importantly, patients often do not present with the typical triad, but may exhibit isolated encephalopathy, unexplained visual disturbance or hearing loss, delaying the diagnosis or resulting in misdiagnosis. 3-5 Rapid diagnosis is, however, essential to ensure early immunosuppressive therapy. Cerebral magnetic resonance imaging (MRI) and retinal fluorescein angiography play an important role in confirming the diagnosis.
Recently, all reported cases were reviewed and it was reported that resolution of abnormalities as demonstrated by T2 MRI is rare. 2 We now report four consecutive cases; in three patients treatment was initiated relatively rapidly with a favourable clinical outcome, with clearance of T2 MRI lesions, suggesting that early treatment is associated with resolution of abnormalities.
In addition, in diseases such as Susac syndrome it is often unclear which doctor is responsible for disease monitoring and gathering all the information to take treatment decisions. We suggest that the different specialists involved cooperate, and that treatment decisions are guided by a rheumatologist or immunologist.
CASE REPORTS
Case 1
A 38-year-old woman presented in October 2009 with intermittent paresthesias and numbness of her left hand and left cheek, diminished memory and reduction of vigilance. Physical examination was normal except for mild memory deficits. Routine laboratory studies were normal. Cerebrospinal fluid analysis showed 13/3 cells/ul, protein 1.71 g/l, without oligoclonal bands. Cerebral MRI showed multiple symmetrical white matter lesions in the centrum semiovale, corpus callosum, left thalamus, left pons and right cerebellar peduncle. The aspect and distribution of these lesions favoured the diagnosis of multiple sclerosis (MS). One week later, the patient was admitted because of progressive cognitive impairment. Initial ophthalmological evaluation was normal. Repeated brain MRI showed rapid progression of the white matter lesions. The differential diagnosis consisted of acute disseminated encephalomyelitis (ADEM) or primary cerebral vasculitis. She was treated with pulse methylprednisolone 1000 mg/day for five consecutive days, after which cognitive function improved.
Three weeks after presentation, the patient complained of blurred vision and a black spot in the left eye. Fundoscopy showed abnormalities suspect for vasculitis in both eyes. Under the working diagnosis of cerebral and ocular vasculitis, treatment with prednisolone 1 mg/kg/day and azathioprine 100 mg/day was initiated.
In the beginning of December, she presented with ataxia, vertigo, and vomiting. Fluorescein angiography revealed BRAOs in both eyes, confirming the diagnosis of Susac syndrome. In retrospect, BRAOs were also present one month earlier. Methylprednisolone 1000 mg/day was administered for three days. A few days later, tinnitus and perceptive hearing loss of the right ear occurred. Prednisolone was increased to 80 mg/day, a five-day course of intravenous immunoglobulins (IVIG) was prescribed, and aspirin was added. Consecutively, she started with monthly cyclophosphamide infusions.
In January 2010, she was retreated with methylprednisolone 1000 mg/day for five days because of progressive ataxia, headaches, paresthesias and memory loss. In March 2010 an additional IVIG course was given because of progressive visual loss caused by new BRAOs.
In June 2010, cognitive impairment prevented the patient from working, reading, writing, household activities and hobbies. Fluctuating perceptive hearing loss in both ears required hearing devices. Two infusions of rituximab 1000 mg 2 weeks apart were given. Cyclophosphamide was continued as maintenance therapy once every three months until March 2011, and the prednisolone was gradually tapered and finally discontinued in June, 2011. No new BRAOs had occurred.
In September 2010 she complained of feeling increasingly depressed and was referred to a psychiatrist. A depressive disorder was diagnosed, most likely as a consequence of the severe disability, possibly secondary to central nervous system damage. Her depressive complaints improved with sertraline, and she was referred for further neurological and cognitive revalidation and rehabilitation. She retained severe neuropsychological sequelae (memory problems and disturbed executive capacities). Follow-up MRI studies in October 2010 and June 2011 showed a slight decrease in lesion load and volume with discrete parenchymal defects in previously active lesions, without active or new lesions.
Case 2
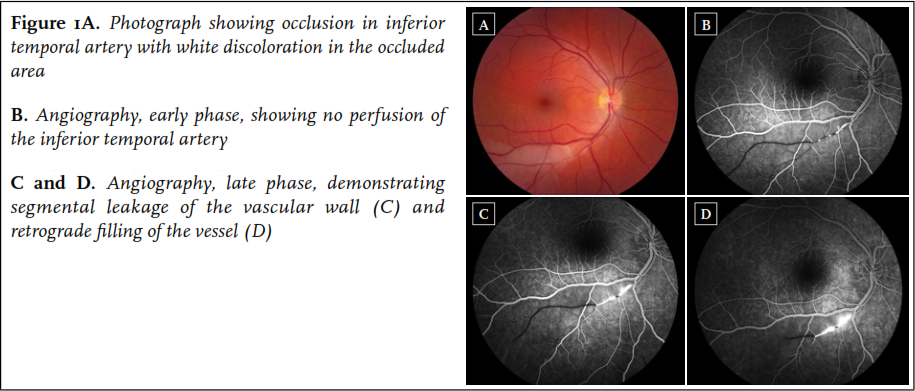
A 34-year old woman first presented in August 2010 with sudden occurrence of a black spot in the right eye. She had a one-year history of progressive migraine-type headaches with visual aura, and Raynaud’s phenomenon of the hands. Physical examination and routine laboratory studies were normal. Fluorescein angiography revealed a BRAO of the right eye ( figure 1). Cerebral MRI was performed, revealing multiple lesions in the corpus callosum, including a typical snowball lesion. In addition, a few white matter lesions and some lesions in the right frontal lobe were noted. Audiometry was normal. Based on the typical MRI findings combined with BRAO, the diagnosis of Susac syndrome was made.
The patient was treated with methylprednisolone 1000 mg/day for three consecutive days, followed by oral prednisolone 1 mg/kg/day, and aspirin. Mycophenolate mofetil 2dd 1000 mg was initiated because of fertility concerns related to cyclophosphamide use. She responded well to treatment and the prednisolone was gradually tapered to 10 mg/day after five months and eventually discontinued in June 2011 because of remission. She was functioning normally and had started working again. Follow-up cerebral MRI after one year revealed discrete resolution of white matter lesions, and a parenchymal defect on the site of the initial snowball lesion in the corpus callosum.
In August 2011 mycophenolate mofetil was switched to azathioprine 150 mg/day because of anaemia and a wish to become pregnant. In June 2012, recurrence of BRAO in the left eye and subtle cognitive dysfunction required reinstitution of the mycophenolate mofetil and corticosteroids. In October 2012, azathioprine was started again because of a pregnancy wish and, while pregnant, azathioprine was discontinued in July 2013 because of anaemia. In August 2013, a healthy daughter was born by caesarean section. Six weeks after delivery, however, she presented with a relapse of disease activity including mild hearing loss of the right ear, visual auras, and a new BRAO with leakage of fluorescein. Prednisolone was increased to 60 mg/day and the azathioprine was restarted. She responded very well with full recovery of her symptoms.
Case 3
A 39-year-old woman presented to the emergency ward of another hospital in August 2012 with vertigo, gait ataxia, headaches with photopsia, nausea and vomiting. Medical history was unremarkable except for familial hypercholesterolaemia for which she had been using statin therapy since 1994. Three days after presentation, she developed a bilateral perceptive hearing loss of 30-35 dB. She also experienced transient visual symptoms, central facial paresis, and paresthesias in the right arm and cheek. Cerebrospinal fluid analysis showed 8 cells/ul, and protein of 2.34 g/l. Cerebral MRI showed T2 hyperintense lesions in the basal ganglia and posterior fossa, as well as leptomeningeal enhancement on post-gadolinium T1; no abnormalities were observed in the corpus callosum. The ophthalmologist diagnosed a trochlear nerve paresis of the right eye. Differential diagnoses included infectious causes as well as neurosarcoidosis. Reinvestigation by the ophthalmologist provided the clue to the diagnosis 2.5 weeks after presentation, when BRAOs with typical segmental fluorescein leakage proximal of the occluded areas were visualised in both eyes, confirming the diagnosis of Susac syndrome. The patient was transferred to our hospital. Her symptoms had improved in the meantime, with only a slight gait ataxia and a visual field defect in the right eye.
Three weeks after the initial presentation, treatment was started with methylprednisolone 500 mg/day for five days, followed by oral prednisolone 1 mg/kg/day. Azathioprine 150 mg/day was added. Further clinical improvement in the next weeks enabled the prednisolone to be gradually tapered to 10 mg/day in April 2013. Ophthalmological evaluation had improved without new BRAOs, audiometry confirmed mild stable perceptive hearing loss especially of the left ear. Repeated MRI of the cerebrum after seven months was completely normal, all the T2 hyperintense lesions in the posterior fossa as well as leptomeningeal enhancement had disappeared. After one year, the prednisolone was discontinued, and the patient continued on azathioprine 150 mg/day.
Case 4
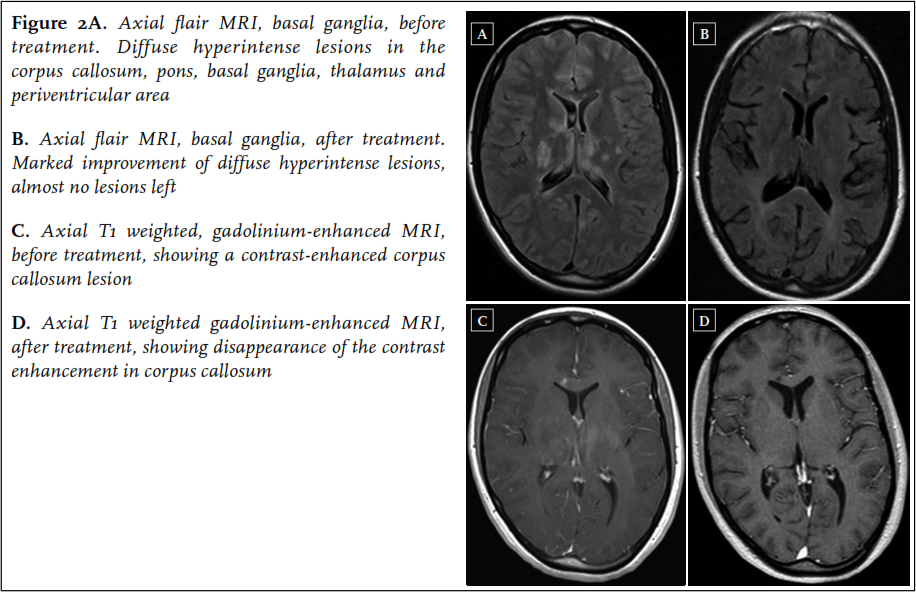
A 22-year-old woman was admitted to the neurology ward of another hospital in the beginning of January 2013. In the past ten weeks, she had experienced several episodes starting with paresthesias in the left arm and leg, followed by a right-sided headache, initially diagnosed as migraine with aura. In the weeks before admission the headaches became more severe, together with occurrence of a progressive gait disorder, fatigue and memory loss. On neurological examination at admission, she was bradyphrenic and bradykinetic, and had a decreased sensation, hyperreflexia and a grade 4 paresis of the left arm and left leg. Short-term memory was impaired. Laboratory tests were normal. Cerebrospinal fluid analysis showed a lymphocytic pleocytosis with 4 cells/ul and a total protein of 2.29 g/l. Brain MRI revealed multiple hyperintense lesions on the FLAIR and T2-weighted images in the corpus callosum, pons, basal ganglia, thalamus and periventricular area, not typical for MS ( figure 2). The next day, sudden severe perceptive hearing loss of the right ear occurred, followed by acute visual loss in the right eye the day thereafter. Ophthalmological evaluation revealed BRAOs in the right eye. A diagnosis of Susac syndrome was made. The patient was treated with two three-day courses of methylprednisolone 1000 mg/day one week apart, followed by oral prednisolone 60 mg/day, and aspirin. In addition, mycophenolate mofetil 2dd 1000 mg was started. Soon after commencing treatment, the left-sided neurological symptoms resolved, although some small new BRAOs did occur in the first weeks after treatment initiation. The deafness of the right ear persisted. She was referred to a rehabilitation clinic.
In May 2013, further clinical improvement was noted: the right ear showed a small perceptive loss, no new BRAOs occurred, although a persistent visual field defect in the upper part of the right eye remained. Follow-up MRI of the brain showed a dramatic improvement with almost no hyperintense lesions left on FLAIR and T2-weighted images ( figure 2). Prednisolone was tapered to 10 mg/day in August 2013, and mycophenolate mofetil was continued.
DISCUSSION
Epidemiology Most cases of Susac syndrome occur in young women aged 16-40 years, with a mean age of 31.6 years (range 8-65 years). Of the 304 patients recently reviewed by Dorr, 78% were female, which is in line with the presumed autoimmune aetiology of the disease. No racial trends have been detected. 2 True incidence and prevalence is unknown, as the disorder is possibly underdiagnosed.
Aetiology The pathophysiology of Susac syndrome is still unclear; however, an immune-mediated injury involving the endothelium of retina, cochlea and cerebral vasculature is the leading hypothesis. This was suggested by biopsy studies in which endothelial cell necrosis, basement membrane thickening with deposition of collagen, mural and intra-luminal fibrin deposition, and C3d and C4d deposition in capillaries were found. 6 Narrowing or occlusion of microvasculature then results in ischaemic injury of the brain, retina, and cochlea. Similar findings have been reported in muscle and skin biopsy studies in patients with dermatomyositis, suggesting a possible associated disease mechanism. 7,8
Case reports have found anti-endothelial antibodies of IgG1 subclass in patients with Susac syndrome. 9-11 However, these antibodies were also found in patients with Sjogren syndrome and dermatomyositis, and it is unclear if they are involved in the development of endothelial injury, or if they are an unspecific epiphenomenon of the disease.
Disease course
The clinical course of Susac syndrome can be divided into a monocyclic, polycyclic and a chronic continuous course. 5 The monocyclic course is defined as a fluctuating disease that self-limits after a maximum period of two years and does not recur; the polycyclic course is characterised by relapses of disease activity following a period of remission, continuing beyond two years after presentation. A disease-free period of 18 years between two relapses has been described. 12 Dorr and colleagues could classify 54% of 114 patients as monocyclic, 42% as polycyclic, and only 4% (four patients) as having a chronic continuous course, questioning the clinical relevance of this third category. 2 In our four patients, two could not yet be classified due to a follow-up of less than one year, but were in remission; patient 1 had a monophasic course with severe residual symptoms, and patient 2 had a polycyclic course with recurrences after two and three years, with the second recurrence manifesting six weeks after delivery.
Clinical symptoms
The typical clinical triad consists of subacute encephalopathy, visual loss due to BRAO, and sensorineural hearing impairment. It is important to emphasise that only 13% of Susac patients presented with the characteristic triad at disease onset, impeding an early correct diagnosis. In virtually all patients, the full triad will develop during the disease course, after on average five months. 2 Indeed, none of our four patients presented with the classical triad.
The most common clinical manifestations at onset are central nervous system (CNS) symptoms, observed in two-thirds of patients, followed by visual symptoms and hearing disturbances in around 40% of patients at presentation. Migraine-like headaches are reported in 80% of patients at disease onset. 2
Encephalopathic CNS symptoms include cognitive impairment (48% of patients), confusion and/or disorientation (39%), emotional disturbances (16%), behavioural changes (15%), personality changes (12%), apathy (12%), psychosis (10%) and reduction of vigilance (9%). 2 Other CNS manifestations include gait ataxia (25%), vertigo (25%), sensory disturbances (24%), upper motor neuron signs (21%), paresis (20%), nausea and vomiting (15%), dysarthria (13%), oculomotor dysfunction (10%), urinary dysfunction (9%) and diplopia (5%). 2
Reported visual symptoms are black or grey scotomata in the visual field, photopsia, occasionally scintillating scotomata, and visual acuity loss when the central retina is involved. However, patients may also be asymptomatic if only the far periphery of the retina is affected. Visual field loss is permanent.
Hearing loss is irreversible in the majority of patients, and may occur rapidly or develop overnight. Severe hearing loss is often accompanied by vertigo and tinnitus, and may require cochlear implants. Some patients complain about myalgia and/or arthralgia, and might exhibit dermatological signs, such as livedo racemosa. 8,13
It has been suggested that patients who present with encephalopathy are more likely to experience a monocyclic course, and that presentation with visual or hearing impairment without clinically evident encephalopathy is likely to have a prolonged polycyclic course, 2,13 as was the case in our patients 1 and 2, respectively.
Diagnostic procedures
Cerebral MRI, retinal fluorescein angiography and audiometry are considered crucial investigations to enable diagnosis, especially since pathology on these tests has been described more often than symptoms being clinically evident, emphasising an appropriate diagnostic workup. Fluorescein angiography exhibits multifocal non-perfused arterioles (BRAOs) in 99% of patients, 2 highlighting the importance of this investigation. Additional abnormalities include typical segmental fluorescein leakage of the arteriolar wall, and/or yellow retinal arterial wall plaques (Gass plaques). 14
Audiometry reveals perceptive hearing loss in almost every patient, typically involving the low or middle frequencies, because these microvessels are affected first. The evaluation of hearing loss in the encephalopathic patient may be very difficult.
MRI
Cerebral MRI typically reveals multifocal T2-hyperintense lesions of 3-7 mm in diameter most frequently involving the white matter, especially the corpus callosum, periventricular areas, centrum semiovale and subcortical regions. Involvement of the corpus callosum, especially the presence of ‘snowball lesions’ in the centre of the corpus callosum, is considered pathognomonic and was found in all 27 patients with Susac syndrome in a previous MRI study. 15 In two recent reports, however, 21-22% of patients did not have callosal involvement, suggesting that callosal involvement is not mandatory for the diagnosis of Susac syndrome. 2,16 Deep grey matter, basal ganglia and thalamus involvement was described in 70% of patients; cerebellar involvement in 52%; leptomeningeal enhancement in 33%; and brainstem involvement in 30%. 15,17 Over time, atrophy of the corpus callosum, cortex and cerebellum may develop. Sometimes residual holes in the central fibres of the corpus callosum emerge, representing micro-infarctions; these lesions may be pathognomonic.
Resolution of white matter lesions is rare, but has been reported in some studies. 18-20 Interestingly, we observed complete resolution of white matter abnormalities in patient 3, and almost complete resolution in patient 4.
MRI is also helpful in clarifying the differential diagnosis between Susac, MS, and ADEM patients: in MS, corpus callosum lesions are located in the periphery instead of the centre; leptomeningeal enhancement is absent in MS or ADEM, but present in 30% of Susac patients; and deep grey matter involvement seldom occurs in MS but is present in 70% of Susac patients. Kleffner and colleagues suggested that diffusion tensor imaging is superior to conventional MRI, as all patients had disruption of fibre integrity in the genu of the corpus callosum. 21
Additional diagnostic procedures
Additional diagnostic tests are often performed, but are mainly useful for the exclusion of other diagnoses. Routine laboratory measures are usually normal; the erythrocyte sedimentation rate and C-reactive protein may be mildly elevated. The rate of ANA positivity is around 7% and therefore comparable with the healthy population. 2 Anti-endothelial cell antibodies have been detected in the serum of patients with Susac syndrome, 9,10,32 but were reported in other autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, Sjogren syndrome and sarcoidosis as well.
Cerebrospinal fluid analysis is important in patients with CNS symptoms to rule out infectious encephalitis. It usually shows a moderately elevated protein of up to 2 g/l with a mild lymphocytic pleocytosis (5-30 cells/mm3). Isolated oligoclonal bands are rare in Susac syndrome, but are found in up to 98% of MS patients. 23
Cerebral angiography might be helpful in the differential diagnosis of cerebral vasculitis, but is normal in Susac syndrome, since the precapillary arteriole is below the resolution of angiography. Brain biopsy was mainly performed in the older literature, showing focal microangiopathic and gliotic changes in the majority of patients. No overt demyelinisation was reported in any of the cases.
Treatment
No evidence-based standardised treatment protocols exist, as a consequence treatment of Susac syndrome is based on case reports and small case series. 5,7,16 In concordance with the presumed autoimmune endotheliopathic aetiology, treatment has to be immunosuppressive. It seems important that the disease should be treated early, aggressively and long enough to prevent relapses, but the appropriate treatment duration is anecdotal. In the acute phase, pulse methylprednisolone 1000 mg/day for three to five days should be initiated, followed by oral prednisolone 1 mg/kg/day for four weeks. Corticosteroids can then be slowly tapered to 10-15 mg/day after six months, and thereafter to zero in another six months. 24 Additional immunosuppressive medication should be started in the induction phase. Different treatment strategies include cyclophosphamide (monthly infusions of 750 mg/m2), mycophenolate mofetil (2dd 1000 mg), or possibly rituximab (375 mg/m2 once weekly for four weeks). To reduce the risk of thrombosis of the small arterioles, it is recommended to add aspirin in all patients. 7 In severe or refractory cases, IVIG (2 g/kg monthly for six months) or plasmapheresis may be useful. 7,16 Infliximab was reported to give striking improvement in headache and ataxia in a single patient who did not respond to, and experienced side effects from prednisolone. 25
Following induction therapy, maintenance therapy may consist of cyclophosphamide infusions every three months, mycophenolate mofetil, or azathioprine for at least two years. 7
Long-term treatment management is challenging and guided by serial ophthalmological, audiological, neuro psychiatric, and MRI evaluations. Patients with severe hearing loss benefit from cochlear implants.
In all four of our patients, treatment decisions were guided by an experienced rheumatologist as a case manager, reviewing diagnostic studies. All patients were treated with methylprednisolone followed by oral prednisolone. Patient 1 received cyclophosphamide in association with IVIG, and at a later stage rituximab, and cyclophosphamide maintenance treatment every three months; patient 2 received mycophenolate mofetil induction and azathioprine maintenance; patient 3 was treated with azathioprine alone; and patient 4 was treated with mycophenolate mofetil alone.
Prognosis
In the majority of patients, the disease course is monocyclic without relapses after two years. Up to 40% of patients, especially those patients presenting with visual or hearing impairment, experience a polycyclic disease course with remissions followed by exacerbations. While some patients recover without or with minimal sequelae, most patients have residual symptoms despite immuno suppressive treatment, ranging from mild symptoms to severe psychoneurological deficits, hearing loss, and/or visual impairment. 26 In contrast with earlier reports, in our population, three out of four patients had favourable outcomes, possibly reflecting early treatment, although follow-up time is limited in two patients.
CONCLUSION AND FUTURE PERSPECTIVES
In conclusion, we present four new cases of Susac syndrome, illustrating different disease courses and favourable outcomes in three patients. Susac syndrome is an important differential diagnosis in numerous disorders and should be suspected also in case of isolated encephalopathy, visual field defects or hearing loss, as early treatment seems to improve outcomes. To accomplish this, fruitful cooperation between rheumatologist, ophthalmologist, neurologist and ENT specialist is necessary, and we suggest that an immunologist or rheumatologist coordinates treatment decisions. For the future, large prospective cohorts of patients with Susac syndrome are needed to systematically test different treatment regimens and assess outcomes in a standardised way, aiming at better care for patients with this rare but potentially incapacitating disorder.
DISCLOSURES
Funding source: none Conflicting interests: none
REFERENCES