KEYWORDS
Acromegaly, Cushing’s disease, pituitary apoplexy
INTRODUCTION
Pituitary apoplexy, defined as ischaemia or haemorrhage in the pituitary gland, occurs in 10-17% of pituitary tumours.1,2 Clinical presentation is usually characterised by the acute onset of severe headache, visual field defects, meningeal irritation, ophthalmoplegia and hypopituitarism. Apoplexy primarily occurs in macroadenomas and most pituitary apoplexies occur in non-functioning adenomas.3,4 We recently evaluated a case of clinically suspected acromegaly. To our surprise, low insulin-like-growth factor type 1 (IGF-1) levels were detected in this patient. Magnetic resonance imaging (MRI) showed evidence of a recent pituitary apoplexy, which might have led to spontaneous remission of the acromegaly. Furthermore, we recently treated a patient with Cushing’s disease who showed two periods of spontaneous remission after subsequent pituitary apoplexies with disease recurrence in between.
CASE REPORTS
Case 1
A 41-year-old male was referred to our tertiary referral centre in July 2011 with suspected acromegaly because of his distinct facial features and acral enlargement. The patient had a history of diabetes mellitus and insulin therapy since May 2009. He then started losing weight and achieved a weight loss of 24 kg in May 2011. In this period of two years, his blood glucose levels fully normalised and insulin therapy could be stopped. The patient was previously investigated by a maxillofacial surgeon because of dysgnathia due to asymmetrical elongation of the mandibula. The maxillofacial surgeon was the first to suspect acromegaly in this patient.
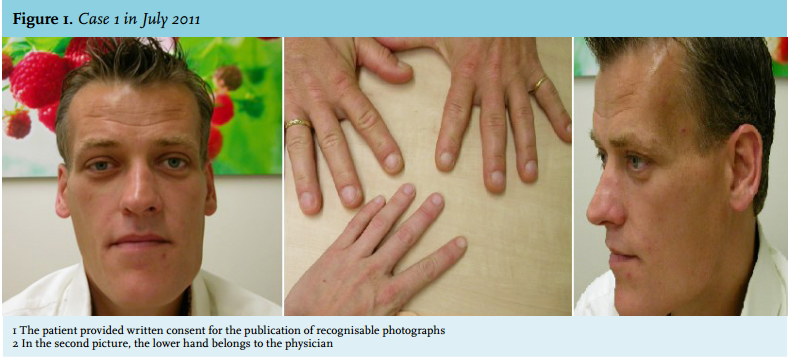
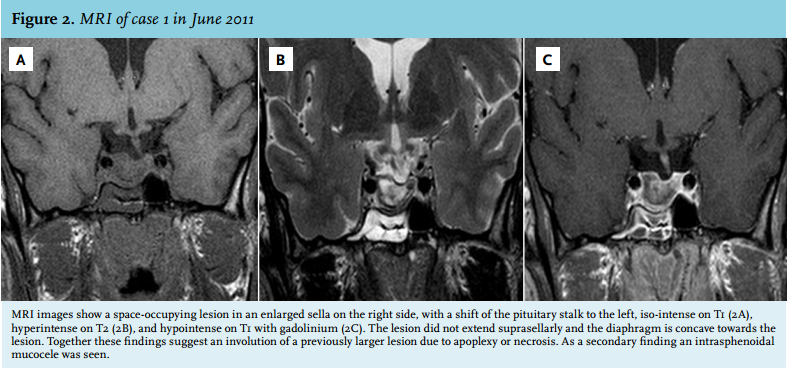
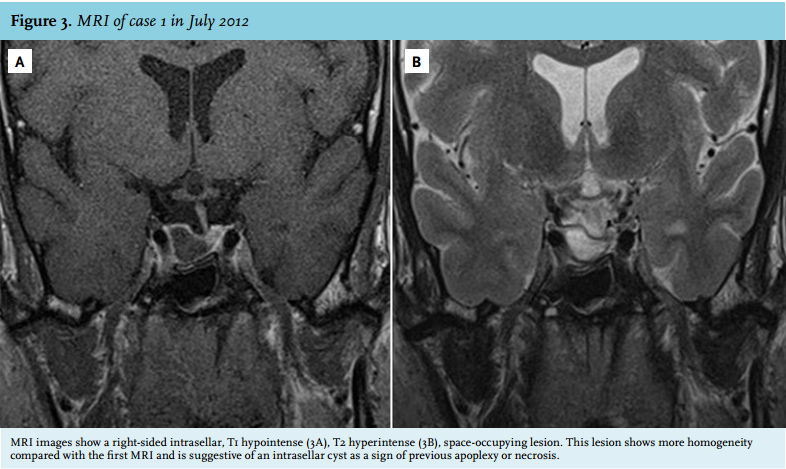
In April 2011, the patient experienced a sudden onset of severe pain in the neck, he did not have any headache. Since then, he complained of fatigue and he felt dizzy after standing up. His facial features had started to change five to ten years before. On physical examination, the acromegalic appearance was striking, characterised by bilateral asymmetric elongation of the mandibula, general asymmetry in facial appearance, the large nose, frontal bossing and large hands (figure 1). However, to our surprise, on laboratory testing he turned out to be growth hormone deficient (IGF-1 3.5 nmol/l (normal value (n) 11.2-32.8 nmol/l), growth hormone peak of 2.4 mU/l during insulin tolerance test (ITT)). In addition, hypogonadotropic hypogonadism was present (testosterone 0.2 nmol/l (n 11-45 nmol/l), follicle stimulating hormone (FSH) 0.74 U/l (n 1.5-11 U/l), luteinising hormone (LH) 0.82 U/l (n 1.4-8.5 U/l) and a partial adrenal insufficiency (cortisol peak during ITT 0.32 µmol/l.) Thyroid function was normal. MRI images revealed a space-occupying lesion in an enlarged sella at the right side, with a shift of the pituitary stalk to the left, iso-intense on T1, hyperintense on T2, and hypointense on T1 with gadolinium. The lesion did not extend suprasellarly and the diaphragm was concave towards the lesion. Together these findings suggest an involution of a previously larger lesion due to apoplexy or necrosis. As a secondary finding an intrasphenoidal mucocele was seen (figure 2). Because of these findings, we hypothesised the previous presence of a growth hormoneproducing pituitary adenoma which was ameliorated by pituitary apoplexy. Repeated MRI imaging in July 2012 showed largely the same picture with signs suggestive of right-sided pituitary apoplexy (figure 3). During follow-up, IGF-1 levels normalised to 13.4 nmol/l (n 9.8-28.6 nmol/l). At this time the testosterone levels increased, but remained low (7.3 nmol/l (n 11-45 nmol/l)). No further dynamic testing of the pituitary adrenal axis has been performed up to now. Further follow-up was planned in order to screen for a possible relapse of the acromegaly.
The pituitary lesion was treated conservatively. The patient was further treated with testosterone and hydrocortisone therapy. The changes in the jaw even led to malocclusion which caused the patient to have eating difficulties. This problem was surgically corrected.
Case 2
The second patient was a 47-year-old female who was admitted to our neurosurgery department in September 2012 with acute severe headache. A computed tomography (CT) scan was performed in order to exclude a subarachnoid haemorrhage. Additional MRI scanning was performed when no subarachnoid haemorrhage was detected. Based on images of the MRI scan, pituitary apoplexy was suspected.
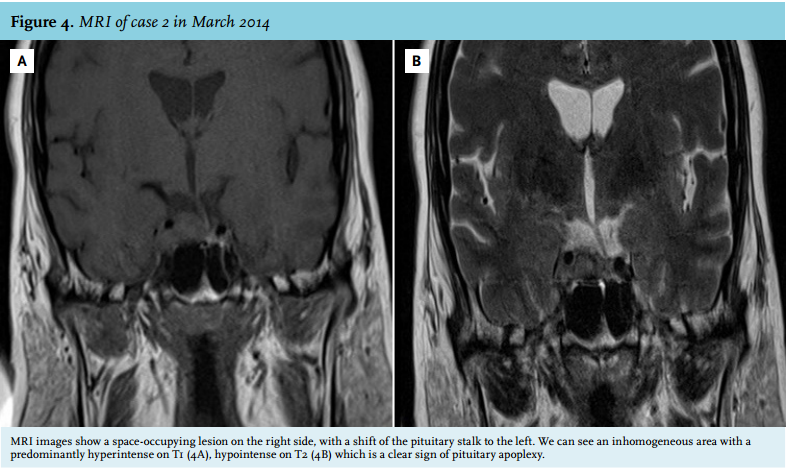
One year before she was diagnosed with diabetes mellitus type 2 and hypertension. Recently, she was analysed for hypercortisolism in another hospital previously to the episode of headache. She reported progressive muscle weakness, fatigue, weight gain of 30 kg in six months, the development of red stretch marks at the abdomen and easy bruising. In August 2012 her cortisol was not suppressed after low-dose dexamethasone (0.96 µmol/l). On physical examination, we noticed central obesity, moon face, supraclavicular fat pads, buffalo hump and abdominal purple striae. Laboratory results showed hypocortisolism (morning cortisol 0.04 µmol/l) and secondary hypothyroidism (free thyroxine 7.1 pmol/l (n 8-22 pmol/l), thyroid stimulating hormone 0.20 mU/l (n 0.4-4.0 mU/l)). The IGF1, LH, FSH and oestrogen levels were within the normal range. MRI images showed a space-occupying lesion at the right side, with a shift of the pituitary stalk to the left and an inhomogeneous area, predominantly hyperintense on T1 (figure 4A), hypointense on T2 (figure 4B).
We hypothesised that this patient had previously suffered from a hormonally active pituitary adenoma which had caused Cushing’s disease, which had now ameliorated after pituitary apoplexy. Hormone substitution therapy was started and the patient was regularly monitored at our outpatient clinic. During follow-up the hydrocortisone dose could gradually be tapered and was eventually stopped in April 2013. At that time, the pituitary-adrenal axis appeared to be fully recovered as was supported by repeated normal midnight salivary cortisol levels and normalised 24-hour urine cortisol levels. In May 2013, the patient started complaining of fatigue again and gained 7 kg of weight. In October 2013, the serum cortisol was not suppressed after low-dose dexamethasone (0.17 µmol/l). However, her urinary cortisol excretion and salivary cortisol were completely normal. At the time of testing the patient was not on any medication that could interfere with the dexamethasone suppression test. The 1 mg dexamethasone suppression test was repeated in February 2014 and the serum cortisol turned out to be 0.37 µmol/l. This was a strong indication that her Cushing’s disease had recurred. Another evaluation of 24-hour urinary cortisol excretion was planned. However, in March 2014, the patient was brought into our emergency department with an acute onset of severe headache and visual impairment. MRI imaging showed a second pituitary apoplexy in the right side of the pituitary characterised by right-sided inhomogeneity, hyperintense on T1, spreading towards the right cavernous sinus (figure 4). After this second episode of pituitary apoplexy, the symptoms of hypercortisolism diminished and the 1 mg dexamethasone suppression test normalised (cortisol 0.04 µmol/l). Because the bleeding did not compress the optic chiasm it was decided not to perform surgery in the acute setting. Instead it was decided to perform elective endonasal endoscopic transsphenoidal surgery in September 2014 in order to remove the haematoma and remaining adenoma tissue to prevent disease recurrence in the future. Pathological examination of the removed tissue revealed typical adenoma tissue with adrenocorticotropic hormone (ACTH) expression.
DISCUSSION
Prior to his referral, the first patient most likely had acromegaly due to a growth hormone-secreting pituitary adenoma. This hypothesis is based on his striking physical appearance and on the MRI findings. Nevertheless, detailed endocrinological evaluation demonstrated apparent cure of acromegaly and even a transient growth hormone deficiency, most likely due to an acute mass effect of the pituitary apoplexy. The clinical findings in pituitary apoplexy vary widely and in this patient this event seems to have occurred ‘silently’ in the period between May 2009 and April 2011.5 Pituitary apoplexy usually occurs spontaneously, as the neovasculature of a tumour is fragile, but it can be associated with a variety of events, including head trauma,6 medications,1 and dynamic pituitary function testing.7,8 However, the exact pathophysiological mechanisms in these associations remain to be elucidated. Interestingly, diabetes mellitus has been associated with infarction of the normal pituitary gland due to its detrimental effects on the microvasculature of the pituitary. Our first patient had diabetes mellitus, which disappeared, possibly due to the remission of acromegaly after pituitary apoplexy. This can be explained by the fact that acromegaly is associated with insulin resistance.9 Substantial lowering of growth hormone levels after pituitary apoplexy has been described before,10 and even spontaneous remission of acromegaly has been reported.11 However, growth hormone deficiency after pituitary apoplexy in acromegaly has never been reported before.
The second patient presented with a remission of Cushing’s disease due to pituitary apoplexy, followed by a period of glucocorticoid deficiency, and a relapse of the Cushing’s disease one year later. Remarkably, six months after the relapse, she again went into remission after a second pituitary apoplexy. Similar cases of remission after pituitary apoplexy followed by relapsing of Cushing’s disease have been described previously.12-14 However, this is the first case report describing a second pituitary apoplexy after relapsing Cushing’s disease. Of note is the short period of glucocorticoid deficiency and remission of just one year before the relapse of Cushing’s disease, which was, together with the prevention of a third apoplexy, a reason to decide to surgically remove the pituitary lesion.
In conclusion, patients in spontaneous remission of hormonally active pituitary adenomas should be suspected of a pituitary apoplexy. Furthermore, even after spontaneous remission after pituitary apoplexy, careful long-term follow-up of these patients is mandatory, as relapses of hormonal hypersecretion can occur.
DISCLOSURES
The authors declare no conflicts of interest. No funding or financial support was received.
REFERENCES